Tumoren des ZNS im Kindes- und Jugendalter – Grundlagen und aktuelle Entwicklungen
CME-Beitrag
Michael C. Frühwald, Johannes Holzapfel, Martin Hasselblatt
Als Entität sind ZNS-Tumoren die häufigsten soliden Neoplasien des Kindes- und Jugendalters. Bis zu 500 Betroffene werden jährlich an die Kompetenzzentren des Deutschen HIT-Netzwerkes (HIrnTumoren) gemeldet. Unter diesen machen die niedriggradigen Gliome annähernd 50% aus. Häufigste bösartige ZNS-Tumoren bei Kindern sind die Medulloblastome. Während die neuroradiologische und -pathologische sowie die Liquor-Diagnostik des HIT-Netzwerkes seit Jahrzehnten in Referenzzentren etabliert sind, nehmen neuere Methoden, wie der Methylierungs-Classifier zur genaueren Einstufung der ZNS-Tumoren, an Bedeutung zu.
In bestimmten Risikokonstellationen kann mit konventionellen Verfahren wie Neurochirurgie, adjuvanter Chemotherapie sowie ggf. Hochdosis-Chemotherapie und Strahlentherapie ein großer Anteil der Kinder und Jugendlichen geheilt werden. Neue Verfahren mittels zielgerichteter Substanzen werden jedoch dringend benötigt, um auch bei Hochrisiko-Neoplasien, wie den malignen Mittellinien-Gliomen, seltenen embryonalen ZNS-Tumoren wie ATRT und ETMR, aber auch metastasierten Erkrankungen bei Säuglingen und Kleinkindern, Fortschritte zu ermöglichen.
Ein besonderer Fokus der Betreuung von Patienten mit ZNS-Tumoren liegt auf der frühzeitigen Erkennung und adäquaten Behandlung von Spätfolgen der Tumorerkrankung, aber auch der tumororientierten Therapie.
Schlüsselwörter: ZNS-Tumoren, HIT-Netzwerk, Methylierungs-Classifier, zielgerichtete Therapien, Spätfolgen
Epidemiologie von ZNS-Tumoren im Kindes- und Jugendalter
Tumoren des ZNS sind bei deutschen Kindern und Jugendlichen die häufigsten soliden Neoplasien [1]. 2009–2013 wurden im Durchschnitt jährlich 480 Kinder und Jugendliche mit ZNS-Tumoren in die Studien des Behandlungsnetzwerks HIT gemeldet (A. K. Gnekow, Erfassung 2018). Sie treten vermehrt bei Jungen auf (Verhältnis männlich : weiblich 1,2 : 1) und neigen zu einer Lokalisation in der hinteren Schädelgrube. Bei Patienten unter zwei Jahren ist die supratentorielle Region mit bis zu 60% die häufigste Lokalisation [2].
Histopathologisch finden sich unter den ZNS-Tumoren bei Kindern und Jugendlichen v. a. niedriggradige Gliome (Low-grade glioma, LGG: 45–50%), gefolgt von Medulloblastomen (ca. 20%), Ependymomen (ca. 10%), Kraniopharyngeomen (ca. 5%) und Keimzelltumoren (ca. 5%; Abb. 1; [2]). Seltenere Entitäten (1–3%) sind Tumoren des Plexus chorioideus (Plexus-Papillome und -karzinome), die atypischen teratoiden/rhabdoiden Tumoren (ATRT) sowie andere seltene embryonale Tumoren, wie die erst kürzlich molekular definierten embryonalen Tumoren mit mehrschichtigen Rosetten (ETMR) und die Pinealis-Tumoren [3].
Die prozentuale Verteilung der verschiedenen Entitäten variiert stark mit dem Alter. So sind ATRT bei Kindern in den ersten sechs Monaten die häufigsten bösartigen ZNS-Tumoren [4]. Zudem nimmt mit steigendem Alter die Inzidenz an Ependymomen, Medulloblastomen und anderen embryonalen ZNS-Tumoren ab. LGG zeigen im Kindesalter einen mehrgipfligen Verlauf mit Höhepunkten um das zweite, vierte bis siebte und zwölfte Lebensjahr [5].
Eine Vielzahl neuer Publikationen zu hochauflösenden molekulargenetischen Untersuchungen hat dazu beigetragen, ZNS-Tumoren weiter zu differenzieren. Insbesondere mithilfe von Methylierungs-Mustern lassen sich viele histomorphologisch kaum unterscheidbare Entitäten in Untergruppen einteilen [6]. Therapieansätze gerade bei Hochrisiko-Neoplasien, wie z. B. den diffus intrinsischen Pons-Gliomen (DIPG) oder auch den H3K27-positiven Mittellinien-Gliomen, orientieren sich nun oft primär an den zugrunde liegenden molekulargenetischen Veränderungen und nicht an der Histomorphologie.

Vom Symptom zur Diagnose
Die Diagnose eines ZNS-Tumors kann altersabhängig komplex und damit auch verzögert sein. So können erste Anzeichen eines ZNS-Tumors mit denen anderer Erkrankungen verwechselt werden, die mit unspezifischen neurologischen Symptomen, wie etwa einer Migräne, einem Torticollis oder Verhaltensauffälligkeiten, aber auch z. B. einer Gastroenteritis einhergehen. Komplizierend kommt hinzu, dass Kleinkinder ihre Symptome in der Regel nur unzureichend beschreiben können, sodass einer ausführlichen und geduldig durchgeführten neurologischen Untersuchung ein nicht zu überschätzender Stellenwert zukommt. Auch unspezifische Symptome sollten die Differenzialdiagnose eines ZNS-Tumors miteinbeziehen [7].
Bei Säuglingen und Kleinkindern kann ein Kreuzen der Perzentilen für den Kopfumfang nach oben (Makrozephalie), ein unerwarteter Gewichtsverlust bis hin zur Kachexie (dienzephales Syndrom), eine gefüllte und/oder vorgewölbte Fontanelle, gesprengte Schädelnähte, prominente Blutgefäße an der Kopfhaut und ein Sonnenuntergangs-Phänomen der Augen hinweisend sein. Diese Symptome sind unspezifische Zeichen einer intrakraniellen Druckerhöhung. Bei älteren Kindern und Jugendlichen stellen lokalisierende Symptome erste Verdachtsmomente dar [7]. Eine Abduzens-Parese oder der Ausfall anderer Hirnnerven kann auf den Befall des Hirnstamms, Krampfanfälle oder kognitive Einschränkungen auf einen Tumor im Bereich der Großhirnrinde hinweisen. Symptome können dabei in ihrer Natur von akut über subakut zu chronisch variieren.
Eine sorgfältig erhobene Anamnese der Beschwerden, einschließlich der bisherigen Entwicklung und des Perzentilen-gerechten oder -fugalen Wachstums, sind zusammen mit der neuropädiatrischen Untersuchung die wichtigsten Schritte auf dem Weg zur Diagnose. Hirnnerven-Ausfälle, Symptome der langen Nervenbahnen sowie eine Ataxie sind z. B. die klassische Symptom-Trias eines Hirnstamm-Prozesses [8].
Notfalltherapie
Ein erhöhter intrakranieller Druck nimmt oft die Gestalt verschiedener klinischer Symptome an (siehe oben). Bei offenen Schädelnähten können Hirndruckzeichen auch lange Zeit komplett fehlen. In den meisten Fällen veranlassen aber hartnäckige, oft morgens schlimmere Kopfschmerzen, (Nüchtern-)Erbrechen, Persönlichkeitsveränderungen und wusstseinsstörungen eine Vorstellung beim Arzt. In der täglichen Praxis führt oft die Feststellung einer Stauungspapille (Papillen-Ödem) zur Überweisung, mit dem Ziel einer weiteren Abklärung, in der Regel mittels Bildgebung des Kopfes.
Eine Reihe konservativer Maßnahmen kann effektiv helfen, einen akut erhöhten intrakraniellen Druck zu senken [2]. Bei vital stabilem Patienten und einem ZNS-Tumor, der ein deutliches perifokales Ödem aufweist, ist eine Therapie mit Kortikosteroiden (z. B. Dexamethason 0,1 mg/kg, max. 60 mg als Kurzinfusion, gefolgt von einer 3–4-mal täglichen Gabe, max. 4 x 8 mg) oft alleine ausreichend, um eine Stabilisierung zu erzielen. Hinweise, dass Kortikosteroide Tumorzellen gegen Chemotherapie resistent machen können, haben ihren Gebrauch (v. a. beim klinisch stabilen Patienten) deutlich eingeschränkt [9]. In Notfallsituationen bei vital bedrohlichen Zuständen, z. B. aufgrund eines deutlichen Hirnödems oder auch einer Liquor-Zirkulationsstörung, sollte gemeinsam mit den Kollegen der Neurochirurgie die Indikation zur unmittelbaren, externen Liquor-Drainage diskutiert werden. Gleiches gilt für eine möglichst frühe operative Tumor(teil-)exstirpation. Während stets die Möglichkeit einer kompletten Tumorentfernung zu prüfen ist, muss in der Nutzen-Risiko-Analyse das mögliche Auftreten irreparabler neurologischer Funktionsausfälle infolge einer radikalen Operation gegenüber der Akuität und den potenziellen Spätfolgen der ZNS-Tumorerkrankung abgewogen werden. Weitere konservative Schritte wie Glyzerin-Infusionen, Hyperventilation zur CO2-Senkung, die Hypothermie sowie Mannit und andere osmotische Diuretika sind der Intensivstation vorbehalten.
Rückenschmerzen mit lokalisierter Schmerzempfindlichkeit treten bei 80% der Patienten mit Rückenmarkskompression auf (spinale Tumoren oder Metastasen). Wegen des Potenzials für bleibende neurologische Schäden sowie der oft ausgeprägten Symptomatik ist es wichtig, die Behandlung unmittelbar zu beginnen, z. B.:
1) Dexamethason vor der definitiven Diagnose;
2) Notfall-MRT mit und ohne Gadolinium-Kontrast nach der Anbehandlung mit Dexamethason;
3) Wenn eine spinale Raumforderung gesichert wurde, muss rasch eine Dekompression folgen (z. B. Laminotomie). Erste Schritte in der Behandlung von Krampfanfällen sind die Sicherung lebenswichtiger Funktionen und – bei Bedarf – rasch die Gewährleistung einer therapeutischen Blutkonzentration eines antiepileptischen Arzneimittels [2].
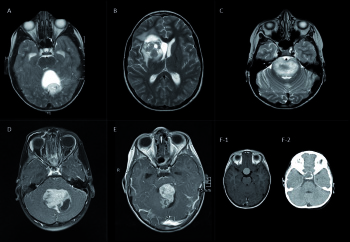
Staging mit bildgebenden und zytologischen Verfahren
Abgesehen von Notfällen und zur Darstellung von Tumor-assoziiertem Kalk (z. B. Kraniopharyngeome; Abb. 2F) sollte die kranielle Computertomografie (cCT) aufgrund der deutlich höheren Strahlenbelastung als einem Kernspintomogramm (MRT) nachrangig eingeordnet werden. Die kraniospinale MRT dient sowohl der Diagnose als auch dem Staging und der differenzialdiagnostischen Abgrenzung zu intrakraniellen/intraspinalen Raumforderungen anderer Ätiologie (z. B. Infektionen; Beispiele in Abb. 2). In einzelnen Fällen (v. a. bei ATRT) ist die Durchführung einer Ganzkörper-Untersuchung indiziert (synchrone Tumoren oder Metastasen außerhalb des ZNS möglich). Sollte eine Ganzkörper-MRT nicht möglich sein, kann man versuchen, durch PET-CT (Positronen-Emissions-Tomografie) die Ausbreitung der Erkrankung zu erfassen.
Da die Standard-MRT-Protokolle für Gehirn und Rückenmark zwischen verschiedenen radiologischen Instituten variieren, wurden Empfehlungen für die vereinheitlichte Bildgebung erstellt [10]. Zum Nachweis oder Ausschluss einer meningealen Beteiligung sind sagittale T1-gewichtete Sequenzen nach KM erforderlich. Die sagittale Schichtdicke darf 3 mm nicht überschreiten. Die physiologischen Venen des Rückenmarkes können mit Knoten einer Meningeose verwechselt werden, hier sind lückenlose axiale Schichten (Schichtdicke kann individuell gewählt werden) für alle verdächtigen Bereiche unerlässlich.
Neben der Erfassung von Metastasen in der Bildgebung muss in jedem Fall eine Untersuchung des lumbalen Liquors erfolgen. Der ideale Zeitpunkt hierfür ist die Untersuchung 14 Tage nach der diagnostischen Tumoroperation. Die Einteilung der Tumorausdehnung folgt dem Chang-Staging [11]. Dieses kombiniert die zytologischen und MRT-Befunde und erlaubt eine Gradierung. Sollte die Lumbalpunktion früher erfolgen und sollten keine Zellen nachweisbar sein, so ist eine erneute postoperative Punktion hinfällig, sollte sie jedoch positiv ausfallen, muss erneut 14 Tage postoperativ punktiert werden. Dies ist nötig, um eine echte Liquor-Dissemination von OP-bedingten Artefakten (z. B. Makrophagen und aktivierte Mono- und Lymphozyten) abzugrenzen.
Weitere bildgebende, z. B. nuklearmedizinische Verfahren wie das FET-PET sind im Kindes- und Jugendalter bislang nicht systematisch untersucht, sind aber wie bei Erwachsenen auch vielversprechend, gerade im Bereich der gezielten Therapien [12].
Multidisziplinarität als Schlüssel zur langfristigen Betreuung
Die Diagnose eines ZNS-Tumors im Kindes- und Jugendalter bedeutet eine außerordentliche Belastungssituation für die betroffenen Patienten, aber auch für ihre Familien. Sowohl die diagnostischen Schritte als auch die therapeutischen Erwägungen und schließlich die Langzeitbetreuung erfolgen in einem multidisziplinären Team, das sowohl medizinische Expertise (Neuropädiatrie, Kinderonkologie, Neurochirurgie, Strahlentherapie, Nuklearmedizin und Neuroradiologie) als auch die Pflege und ein psychosoziales Team beinhaltet. In den Empfehlungen des gemeinsamen Bundesausschusses (G-BA) zur stationären Versorgung von Kindern und Jugendlichen ist präzise festgelegt, welche Qualitätsanforderungen in der Betreuung Betroffener anzulegen sind. Die Erfüllung dieser Kriterien ist Voraussetzung für die Zertifizierung eines Zentrums im Modul Kinderhämatologie und -Onkologie der Deutschen Krebsgesellschaft (OnkoZert).
Die Diskussionen innerhalb des multidisziplinären Teams führen zu einer Empfehlung bezüglich Art und Invasivität der therapeutischen Maßnahmen von der Chirurgie über die Chemotherapie bis zur Strahlentherapie. Darüber hinaus wird in regelmäßigen Konferenzen (Tumorkonferenz und interdisziplinäre Teambesprechung) beraten, wie ein Patient in der Nachsorge betreut wird. Gerade bei Kindern und Jugendlichen mit ZNS-Tumoren kommt der Transition in die Erwachsenenmedizin eine wichtige Rolle zu. In vielen Zentren haben sich daher AYA-Gruppen (Adolescents and Young Adults) gebildet. Dies sind Arbeitsgruppen aus Fachleuten der Kinder- und Jugendmedizin sowie der internistischen Onkologie und den weiteren in der Onkologie tätigen Fachdisziplinen.
ZNS-Tumor-Prädispositions-Syndrome
Nach klassischer Sichtweise sind nur etwa ein bis maximal zehn Prozent aller Neoplasien im Kindesalter mit Keimbahnmutationen assoziiert [13]. Neuere Daten deuten darauf hin, dass diese Zahlen sicher höher liegen. Bei Medulloblastomen z. B. waren in einer retrospektiven Analyse fast 20% der Tumoren aus der SHH-Gruppe mit Keimbahnmutationen verknüpft [14]. Die Fachgesellschaft
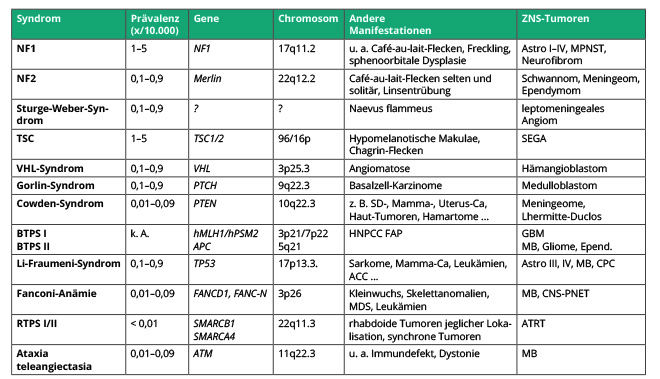
GPOH empfiehlt, alle Kinder und Jugendlichen mit Krebserkrankungen bezüglich Risikofaktoren für das Vorliegen eines hereditären Tumorsyndroms zu überprüfen. Als hilfreich hat sich hierbei eine standardisierte Checkliste erwiesen [15]. In Tab. 1 ist eine Auswahl an Tumor-Prädispositions-Syndromen und der entsprechenden Entitäten sowie der betroffenen Gene wiedergegeben. Beispielhaft sei hier die Neurofibromatose Typ 1 genannt.
Diese ist mit einer Inzidenz von 1 pro 3.500 Neugeborenen das häufigste Tumor-Prädispositions-Syndrom. In bis zu 50% der Fälle finden sich Neumutationen des NF1-Gens in der Keimbahn. Die Diagnose erfolgt meist aufgrund typischer Kriterien. Diese umfassen: ≥ 6 Café-au-lait-Flecken (> 5 mm präpubertär, > 15 mm postpubertär), axilläres, inguinales freckling („skin fold ...“), ≥ 2 Lisch-Knötchen, Sehbahn-Gliom, spheno-orbitale Dysplasie, ≥ 2 Neurofibrome oder ein plexiformes Neurofibrom und ein Verwandter ersten Grades mit NF1. Liegen zwei oder mehr Kriterien vor, gilt die Diagnose als gesichert, und ein standardisiertes Vorsorgeprogramm ist indiziert. Dieses umfasst regelmäßige augenärztliche Untersuchungen (einmal jährlich) sowie klinisch-neurologische Untersuchungen [16]. Regelmäßige kranielle Bildgebung hat sich nicht als hilfreich erwiesen und belastet Patienten und Familien gleichermaßen unnötig.
Eine hervorragende deutschsprachige Ressource für Patienten mit Tumor-Prädispositions-Syndromen und deren betreuende Ärzte findet sich auf der Website www.krebs-praedisposition.de.
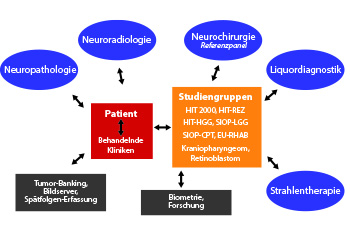
Das Behandlungsnetzwerk „HIT“ der GPOH
Mehr als 90% aller Kinder und Jugendlichen mit einem ZNS-Tumor werden in den deutschsprachigen Ländern in kontrollierten klinischen Studien oder aber auch in Registern (meist als Interimslösung, bevor eine neue Therapieoptimierungsstudie aktiviert werden kann) der Fachgesellschaft GPOH gemeldet. Die Studienleitungen sind gemeinsam mit verschiedenen Referenzinstitutionen im HIT-Netzwerk organisiert. Neben den Kompetenzzentren für verschiedene Entitäten gehören hierzu Referenzzentren für die Strahlentherapie, Neuropathologie, Liquordiagnostik und Neuroradiologie. Hinzu kommen aktuelle Projekte des HIT-Netzwerkes zum Aufbau einer zentralen Hirntumor-Bank, ein zentraler Bildserver, der einen Austausch von Bildmaterial unter strengen Datenschutzvorrichtungen ermöglicht, sowie die Einrichtung eines neurochirurgischen Referenz-Panels (Abb. 3). Eine Herausforderung für das Behandlungsnetzwerk HIT sind die regulatorischen Hürden bei der notwendigen zunehmenden Internationalisierung zur Durchführung von Studien bei kleinen Fallzahlen und beschränkten Ressourcen. Aus einem Deutschen Netzwerk muss so in einem schrittweisen Prozess ein europäisches bzw. internationales Netzwerk für Kinder und Jugendliche mit ZNS-Tumoren entstehen.
Übersicht über aktuelle Entwicklungen bei spezifischen Entitäten
Niedriggradige Gliome (LGG – Low Grade Glioma)
Niedriggradige Gliome der WHO-Grade I und II (LGG) machen mit einer Inzidenz von 10–12/10.000 ca. 45–50% aller primären ZNS-Tumoren im Alter bis zu 15 Jahren aus. LGG sind eine morphologisch heterogene Gruppe von ZNS-Tumoren, die zum überwiegenden Teil durch genetische Veränderungen im MAP-Kinase-Signalweg charakterisiert sind [17]. So weisen annähernd 100% aller pilozytischen Astrozytome (pA) Mutationen dort auf. In 70–80% aller LGG findet sich eine Fusion des Onkogens BRAF mit dem 5‘-Lokus des KIA1549-Gens (KIA1549:BRAF). Neuere Daten belegen mindestens zehn weitere Fusionspartner für BRAF in LGG. Zusätzliche Veränderungen aus dem Signalweg sind BRAF-V600E-Mutationen sowie Läsionen in FGFR1, NTRK2, RAF1 oder KRAS [17].
Bei LGG, die total oder subtotal reseziert werden können und eine Histologie vom Grad I nach WHO präsentieren, ist bei stabilen Befunden eine „Watch & wait“-Strategie gerechtfertigt (Tab. 2). Die möglichst komplette Resektion ist wichtigster Prognosefaktor. Indikationen für eine Chemotherapie – oder aber auch in Abhängigkeit vom Alter eine Radiotherapie – lassen sich klinisch und radiologisch herleiten. Tab. 2 gibt eine Übersicht über Indikationen für den Beginn einer Chemo- oder Strahlentherapie bei nicht-resektablen LGG oder auch Resttumoren. Bei inoperablen LGG wurde bis in die 1990er-Jahre hinein eine Strahlentherapie als Standard angesehen. Aufgrund einer Vielzahl möglicher Spätfolgen bis hin zu radiotherapeutisch induzierten Todesfällen gilt heute die Chemotherapie mit Carboplatin und Vincristin als Standard [18, 19]; die Rolle der Strahlentherapie muss neu definiert werden.
Versuche, die Überlebensraten betroffener Kinder und Jugendlicher durch eine Intensivierung der Chemotherapie zu erhöhen, sind sowohl in Europa als auch USA gescheitert [20, 21].
Säuglinge, insbesondere solche, die einen suprasellären Tumor aufweisen, stellen eine Hochrisiko-Population dar und bedürfen einer neuen Strategie. Ein Alter unter einem Jahr, eine Dissemination sowie das Vorliegen eines dienzephalen Syndroms sind besondere Risikofaktoren [22]. In Vinblastin scheint eine Substanz gefunden, die auch in der Primärsituation Aussicht auf therapeutischen Erfolg bei wenig ausgeprägter Toxizität gibt. Dies gilt insbesondere für Patienten aus der für Nebenwirkungen anfälligen Gruppe der NF1-assoziierten Gliome [23]. In dem bald eröffnenden Protokoll LOGGIC der SIOP werden zum einen unterschiedliche Therapiezeiträume, aber auch die Regimes Vincristin/Carboplatin vs. Vinblastin vs. MEK-Inhibitor (MAP-Kinase-Hemmstoff) gegeneinander randomisiert.
Der primäre Endpunkt in der Therapie von LGG (radiologischer versus klinischer Benefit) muss in jedem einzelnen Fall diskutiert werden. Ein interdisziplinärer Ansatz (Ophthalmologie, Endokrinologie und Neuropsychologie), wie er in vielen Zentren praktiziert wird, muss Standard der Regelversorgung für betroffene Kinder sein. Um der Nachsorge Rechnung zu tragen, wurde in der SIOP-LGG 2004-Studie ein Programm mit regelmäßigen Kontroll-MRT etabliert.
Insgesamt muss man Gliome niedriger Malignität bei Kindern und Jugendlichen als chronische, lebensbegleitende Krankheit mit schubweise auftretendem Wachstum verstehen. Interventionen und Nachsorge müssen entsprechend den Folgen für das tägliche Leben austariert werden [5]. Zukünftige Studien für therapierefraktäre Patienten müssen sich an der Biologie orientieren [17]. Die Frage, ob LGG im Erwachsenenalter eine maligne Entartung erfahren, steht nach wie vor zur Klärung an.

Gliome hoher Malignität (HGG – High Grade Glioma)
HGG sind im Vergleich zu Erwachsenen bei Kindern und Jugendlichen deutlich seltener. Sie machen 3–5% aller Hirntumoren im Kindesalter aus, sind eine histologisch heterogene Gruppe und werden nach der potenziellen Herkunftszelle klassifiziert als astrozytäre Tumoren (anaplastisches Astrozytom (AA), Glioblastom (GBM) und andere, oligodendrogliale Tumoren (anaplastisches Oligodendrogliom) oder oligoastrozytäre (gemischte) Tumoren (anaplastisches Oligoastrozytom). Andere seltene HGG-Typen umfassen anaplastische Gangliogliome, die eine günstigere Prognose als andere HGG zu haben scheinen, und pleomorphe Xanthoastrozytome mit Anaplasie-Zeichen [3]. Eine spezielle Gruppe von HGG ist unter dem Begriff Gliomatosis cerebri (Grad III) zusammengefasst.
Ähnlich wie bei HGG im Erwachsenenalter haben betroffene Kinder und Jugendliche mit einem medianen Überleben von nur 9–15 Monaten außerordentlich schlechte Heilungschancen [24, 25]. Genomische Analysen belegen, dass sich HGG im Kindesalter (pHGG) deutlich von denen erwachsener Patienten unterscheiden [26, 27].
Aktuell zeichnen sich anhand molekulargenetischer Analysen fünf verschiedene Subgruppen von pHGG ab, die v. a. durch somatische Mutationen in Varianten von Histon H3 charakterisiert sind. Die WHO-Klassifikation hat dies entsprechend gewürdigt und definiert eine Gruppe von diffusen Mittellinien-Gliomen anhand der Mutationen in H3K27 (H3.3K27m und H3.1K27m; [3]). Die Heilungsaussichten betroffener Kinder und Jugendlicher in dieser Gruppe liegen bei annähernd 0% [24]. Im Rahmen der kürzlich abgeschlossenen HERBY-Studie wurde geprüft, ob das Hinzufügen von Bevacizumab zu Temozolomid und Strahlentherapie die Prognose verbessern kann. Die Ergebnisse waren insgesamt enttäuschend; jedoch konnte die retrospektive Analyse der molekularen Daten und die Korrelation mit den Therapiedaten helfen, pHGG noch weiter zu differenzieren [25, 28]. So waren pleomorphe Xanthoastrozytome von CD8-Lymphozyten infiltriert und sollten damit gute Kandidaten für eine Therapie mit Checkpoint-Inhibitoren sein. Interessanterweise fanden sich auch vier Adoleszente mit IDH1-Mutationen mit einer verbesserten Prognose. Diese repräsentieren vermutlich die unterste Altersgruppe einer Kohorte von Erwachsenen-Gliomen, sodass diese Patienten in Zukunft in Studien in entsprechend molekular definierten Gruppen stratifiziert werden sollten. Bislang unklar ist, weshalb Säuglinge insgesamt bessere Therapieresultate erzielen [29].
Während die Rolle von Neurochirurgie und Radiotherapie bei pHGG gut etabliert ist, stehen eindeutige Belege für den Nutzen einer adjuvanten Therapie aus. Die HIT-HGG-Studie prüft im HIT-HGG-2013-Konzept randomisiert die Frage, ob der Histondeacetylase-Inhibitor Valproinsäure oder Chloroquin als Autophagie-Inhibitor zusätzlich zu Radiotherapie und Temozolomid das Überleben verbessern können. Weitere zukunftweisende Therapiestrategien für pHGG könnten Immuntherapien wie CAR-T-Zellen oder Kombinationsstrategien mit Checkpoint-Inhibitoren sein [30, 31].
Diffus intrinsische Pons-Gliome (DIPG)/Maligne Pons-Tumoren
Tumoren des Hirnstammes umfassen bis zu 10–20% aller ZNS-Tumoren bei Kindern im Alter von 0–16 Jahren; die meisten davon sind histologisch Gliome [8]. Vor der Ära der modernen bildgebenden Verfahren galten alle „Hirnstamm-Gliome“ als eine einheitliche pathologische Entität und die Prognose als universell infaust. Verschiedene Bildgebungsverfahren wurden angewendet, um Hirnstamm-Tumoren auf Grundlage von Wachstumsmuster und chirurgischer Resektabilität zu klassifizieren. DIPG sind radiologisch als Raumforderungen definiert, die mitten in der Pons liegen, mehr als 50% von deren anatomischer Struktur einnehmen und diese dadurch aufweiten [8]. Trotz einer enormen Vielzahl an Studien und verschiedenster Therapieansätze sterben annähernd alle Betroffenen zwischen sechs Monaten und maximal zwei Jahren nach der Diagnose. Die weitreichende Infiltration und überwiegend hochgradig malignen Eigenschaften dieser Tumoren, gepaart mit ihrer kritischen Lage und Inoperabilität führten auch in der Studie HIT-HGG-2007 zu einem extrem ungünstigen Verlauf [32]. Für experimentelle Therapieansätze wie z. B. den EGFR-Antikörper Nimotuzumab oder auch die Konvektions-verstärkte lokale Infusion von Carboplatin in die Pons konnten allenfalls marginale Effekte belegt und letztlich kein Langzeit-Überlebensvorteil gezeigt werden [33, 34]. Neuere molekulargenetische Daten, die nur durch konsequentes Biopsieren dieser Hochrisikotumoren gewonnen werden konnten, belegen, dass der WHO-Grad allein nur ein unzureichender prognostischer Marker für DIPG ist [35]. Wie bei den pHGG konnten auch in bis zu 85% aller DIPG epigenetische Mutationen wie z. B. H3K27m nachgewiesen werden [36]. Darüber hinaus fanden sich Mutationen in ACVR1, die auch therapeutisch angegriffen werden können [37]. Diese Daten haben das Denken über DIPG verändert. So ist die Diagnose „DIPG“ sowohl eine radiologische als auch eine klinische. Maligne Tumoren der Pons dagegen sind histologisch und molekulargenetisch definiert [8]. Während sich die beiden Begriffe überlappen, sind sie nicht synonym zu gebrauchen.
Nach wie vor bleiben Radiotherapie und ggf. auch Re-Bestrahlung die wichtigsten Stützen der Behandlung [38]. Eine Biopsie sollte nur im Rahmen von klinischen Studien erfolgen.
Medulloblastome (MB)
Medulloblastome (MB) und andere embryonale Tumoren, wie die erst kürzlich molekular definierten ETMR (embryonaler Tumor mit Multilayer-Rosetten), zählen zu den Grad-IV-Tumoren nach WHO [39]. Für diese haben sich zwar in bestimmten Risikokonstellationen multimodale Therapieansätze als erfolgversprechend etabliert, dennoch gibt es sowohl molekular als auch klinisch definierte Hochrisikogruppen (z. B. ETMR, Gruppe 4 MB). Medulloblastome sind die häufigsten bösartigen ZNS-Tumoren bei Kindern und Jugendlichen. Die Entdeckung klinisch relevanter molekular definierter Subgruppen von MB haben die Heterogenität dieser Tumoren bestätigt und neue diagnostische und prognostische Instrumente bereitgestellt.
Die aktuelle WHO-Klassifikation unterscheidet die Untergruppen WNT-, SHH-, Group-3- und Group-4-Medulloblastome [3]. Während WNT-Medulloblastome durch eine exzellente Prognose und in den allermeisten Fällen das Fehlen von Metastasen gekennzeichnet sind, lassen sich die SHH-Medulloblastome in solche mit günstiger und andere mit ungünstiger Prognose einteilen [40]. Mit der Studie SIOP PNET 5 MB wurde eine neue Ära klinischer Studien eingeleitet, in der erstmals molekulargenetische Informationen (CTNNB-Mutationen) in die Risikostratifizierung Einzug gefunden haben. In PNET 5 wird eine Reduktion an Therapieintensität für Medulloblastome der WNT-Gruppe mit niedrigem Risiko evaluiert und im Standardarm die simultane Gabe von Carboplatin anstelle von Vincristin zur Radiotherapie geprüft. Die neuen molekulargenetischen Daten zu einer Hochrisiko-Gruppe von SHH-Tumoren, aber auch zu solchen der WNT-Gruppe mit Hochrisiko-Faktoren (> 16 Jahre, Resttumor, Metastasen), haben zur Einführung einer Therapiestrategie für Hochrisiko-Neoplasien der entsprechenden molekulargenetisch definierten Subgruppen geführt.
Eine besondere Risikogruppe sind Kleinkinder und Säuglinge mit Medulloblastom. In den aktuellen Plänen der internationalen Konsortien werden Patienten der Gruppe-3/4-MB intensiviert behandelt (z. B. mit Hochdosischemotherapie-Ansätzen, Studie SIOP Young Children MB).
Ependymome (EP)
Ependymome weisen je nach ihrem anatomischen Ursprungsort spezifische genetische und epigenetische Veränderungen sowie Expressionsmuster auf. Auf der Basis epigenetisch definierter Faktoren lassen sich neun konsistente Untergruppen (je drei pro anatomischer Region – supra-, infratentoriell und spinal) definieren (ST-SE, ST-EPN-YAP1 und
-RELA, PF-SE, PF-EPN-A und -B sowie SP-SE, SP-MPE und SP-EPN; [41]). Dieses Klassifikationsschema weist bezüglich der Risikostratifizierung Vorteile gegenüber der ausschließlich morphologischen Gradierung auf, sodass nach einer Konsensus-Empfehlung die Therapie von Ependymomen nicht nach WHO-Grad alleine, sondern nach einer integrierten neuropathologisch-molekulargenetischen Diagnose erfolgen soll [42]. Patienten mit supratentoriellen Ependymomen der Gruppe ST-EPN-YAP1 und solche mit infratentoriellen Tumoren der Kohorte PF-EPN-B haben recht gute Heilungschancen, sodass es gerechtfertigt ist, für diese Patienten deeskalierte Therapieschemata anzuwenden. Der Stellenwert von Radiotherapie sowie adjuvanter Chemotherapie nach kompletter Resektion steht hier zur Überprüfung an. Patienten mit Hochrisiko-Tumoren wie z. B. aus der Gruppe ST-EPN-RELA werden aktuell nach wie vor intensiv und multimodal behandelt. Der Grad der initialen Tumorresektion und die lokale Bestrahlung bleiben hier stärkste prognostische Faktoren [43]. Eine Re-OP muss bei Resttumoren grundsätzlich diskutiert werden. Mit modernen Operations- und Bestrahlungstechniken sollen die langfristigen kognitiven und funktionellen Schäden möglichst minimiert werden. Die Rollen der adjuvanten Chemotherapie sowie des Histondeacetylase-Inhibitors Valproinsäure werden in der europäischen „Umbrella“-Studie SIOP EPENDYMOMA II evaluiert.
Keimzelltumoren des ZNS (ZNS-GCT)
ZNS-GCT lassen sich vereinfachend in Germinome (ca. 60% der ZNS-GCT) und nicht-germinomatöse Keimzelltumoren (NGGCT) differenzieren. Die NGGCT umfassen Embryonalkarzinome, Dottersack-Tumoren (oder endodermale Sinuszell-Tumoren), Chorion-Karzinome, reife und unreife Teratome und Teratome mit maligner Transformation sowie gemischte Keimzelltumoren [44]. Die Terminologie der „nicht-germinomatösen Keimzelltumoren“ ist immer wieder infrage gestellt worden, da der Begriff die Vielfalt an Tumoren nicht sicher komplett erfasst. Viele NGGCT (Pathologie enthält mindestens einen NGGCT-Subtyp) sind in ihrer Natur gemischt und beinhalten möglicherweise auch Komponenten eines Germinoms. Daher befürworten einige Autoren den Begriff „gemischte maligne Keimzelltumoren“ (MMGCT; [45]).
Eine platinbasierte Chemotherapie sowie die Strahlentherapie sind sowohl für Germinome als auch für die NGGCT als Standard anzusehen [46, 47]. Ziel der 2011 initiierten Studie SIOP CNS-GCT II ist es, sowohl pathologische als auch genetische Unklarheiten zu beseitigen sowie Behandlung und Prognose der Patienten zu verbessern. Lokalisierte und bifokale Germinome erhalten eine stratifizierte kombinierte Behandlung mit Carboplatin-basierter Chemotherapie (Carboplatin/Etoposid und Etoposid/Ifosfamid im Wechsel) und ventrikulärer Bestrahlung; bei metastasierten Germinomen erfolgt eine kraniospinale Bestrahlung. Bei den NGGCT wird eine dosiseskalierte Chemotherapie, kombiniert mit Bestrahlung, eingeführt. Hochrisiko-NGGCT (AFP > 1.000 ng/ml oder Alter < 6 Jahre) werden weiter eskaliert und erhalten HD-PEI (Eskalation von Etoposid und Ifosfamid).
Kraniopharyngeome
Kraniopharyngeome gehen von Plattenepithelzellen aus, die nach teilweiser Involution der Rathke-Tasche bei der Bildung der vorderen Hypophyse verbleiben. Von Natur aus sind sie langsam wachsende, schleichende intra- und supraselläre Tumoren, die durch lokale Ausbreitung und Schädigung der benachbarten Hypophyse, des Hypothalamus und der Region des III. Ventrikels oder durch erhöhten intrakraniellen Druck oder einen Hydrozephalus Symptome verursachen [48]. Kraniopharyngeome können prinzipiell in jedem Alter auftreten, präsentieren sich aber schwerpunktmäßig mit einer zweigipfligen Verteilung mit Höhepunkten in der Kindheit sowie im Erwachsenenalter. Histologie, Symptomatik und der klinische Verlauf der pädiatrischen Kraniopharyngeome unterscheiden sich in vielerlei Hinsicht von der Variante bei Erwachsenen. Vordringlich ist im Kindesalter das Abwägen potenzieller schwerwiegender Akut- und Spätfolgen durch die Therapie gegenüber der Bedrohung durch den Tumor selbst [49].
Die optimale Behandlung dieser Tumoren bleibt umstritten. In vielen Fällen ist bei hohem Kalkanteil eine komplette chirurgische Exstirpation nicht möglich, sodass eine Strahlentherapie notwendig wird. Die Strahlentherapie, einschließlich Techniken wie Stereotaxie, konformaler Bestrahlung und Protonentherapie, aber auch zielgerichtete Chemotherapie-Ansätze (z. B. Interferon bei solitären Zysten) haben eine Krankheitskontrolle dieser Tumoren über längere Zeiträume ermöglicht [50].
Neben einer Überwachung der endokrinen Folgen des Tumors und der Erkrankung selbst (z. B. Panhypopituitarismus) stehen psychosoziale Spätfolgen im Vordergrund (Adipositas und deren Folgen, kognitive Beeinträchtigungen). Folgerichtig wird im Rahmen der geplanten Studie „Kraniopharyngeom 2017“ eine Randomisierung von Interventionsstrategien bezüglich dieser beiden Dimensionen (Adipositas-Prävention und kognitive Interventionen) durchgeführt.
Atypische teratoide/rhabdoide Tumoren (ATRT)
ATRT sind seltene embryonale ZNS-Tumoren, die in Deutschland nur mit ca. 15–20 Fällen jährlich auftreten. Allerdings sind sie bei Säuglingen unter sechs Monaten die häufigsten bösartigen ZNS-Tumoren und stellen betreuende Ärzte vor eine Vielzahl von Herausforderungen (z. B. Metastasierung, synchrone Tumoren, Keimbahnmutationen). Aus den Daten von über 350 Kindern mit einem malignen Rhabdoid-Tumor (MRT), die an das europäische Rhabdoid-Register EU-RHAB gemeldet wurden, ergibt sich, dass 80% der Betroffenen Säuglinge und Kleinkinder in den ersten beiden Lebensjahren sind. Das durchschnittliche Erkrankungsalter für ATRT liegt bei 20–25 Monaten. Sowohl den extra- als auch intraktraniellen rhabdoiden Tumoren
(EMRT und ATRT) sind Mutationen der Chromatin-Remodellierungs-Gene SMARCB1 und (viel seltener) SMARCA4 gemein [4]. Aufgrund der Seltenheit der Erkrankung gab es bisher keine randomisierten Studien.
Im Rahmen der europäischen Studie SIOPE ATRT01 werden erstmals europäische Kinder im Alter zwischen 12 und 36 Monaten randomisiert in einen Standardarm (konventionelle Chemotherapie plus Bestrahlung) oder einen Arm mit einer Triple-Hochdosischemotherapie eingeteilt. In Paralleluntersuchungen werden Fragen zur Lebensqualität und Neuropsychologie in den beiden Armen untersucht.
Seltene Tumoren des ZNS bei Kindern
Nach Daten des Deutschen Kinderkrebs-Registers (DKKR) liegt die Inzidenz aller ZNS-Tumoren bei 29–33 pro 1.000.000 Kinder. Jenseits der oben behandelten häufigeren ZNS-Tumoren gibt es eine große Anzahl sehr seltener oder gar extrem seltener Tumoren, die weniger als 1–5% aller ZNS-Tumoren im Kindes- und Jugendalter ausmachen. Die meisten dieser Entitäten (z. B. ETMR) sind eine diagnostische und therapeutische Herausforderung, da insbesondere aufgrund der niedrigen Fallzahlen verlässliche Daten zu Prognose und Therapie in der Literatur noch fehlen [39].
Wegweisende Forschungsprojekte – INFORM und Neuropath 2.0
Die integrierte neuropathologische und molekulargenetische Diagnostik hat geholfen, ZNS-Tumoren weiter zu differenzieren und neue Entitäten zu definieren [3]. Im Rahmen der einzigartigen Studie Neuropath 2.0 erfolgt die exakte molekulargenetische, radiologische und neuropathologische Aufarbeitung jedes in Deutschland neu diagnostizierten ZNS-Tumors. Im Rahmen einer interdisziplinären Tumorkonferenz (Telefon- oder Videokonferenz) werden die Daten zusammengeführt und auch für kritische Fälle eine Konsensus-Diagnose gestellt. Teilnehmer der Konferenz sind die Studiengruppen-Leiter, die Referenz-Neuropathologen, die Referenz-Neuroradiologen sowie die Spezialisten der molekularen Genetik [51].
In INFORM, einer Studie zu rezidivierten Malignomen des Kindesalters (nicht ausschließlich ZNS-Tumoren), werden potenzielle Strukturen für eine gezielte Therapie mithilfe einer Vielzahl molekulargenetischer Methoden ermittelt [52].
Beide Projekte versprechen, sowohl unser Verständnis für ZNS-Tumoren des Kindesalters grundlegend zu ändern als auch neue Therapiewege zu definieren (s. a. Trillium Krebsmedizin 5/2017, S. 364 ff.).
Spätfolgen
In den letzten Jahrzehnten haben sich die Therapieergebnisse für Kinder und Jugendliche mit ZNS-Tumoren spürbar verbessert. So betrug die 10-Jahres-Gesamtüberlebensrate im Jahre 1980 weniger als 50%, während neuesten Daten des Kinderkrebsregisters (DKKR) zufolge 73% der Kinder mit ZNS-Tumoren länger als zehn Jahre überleben. Diese verbesserten Heilungsraten beruhen sicher ebenso auf der frühzeitigen Einbeziehung der Strahlentherapie wie auf aggressiveren chemotherapeutischen und neurochirurgischen Ansätzen. Parallel zum Überleben nimmt die Inzidenz der langfristigen Spätfolgen zu. Mehr als zwei Drittel der Langzeit-Überlebenden haben mindestens eine chronische medizinische Störung. Zu diesen Folgeerscheinungen gehören Endokrinopathien, Osteoporose, Hirngefäß-Erkrankungen, Dysfunktionen der Sensibilität und Sensorik, Zweittumoren sowie psychische Komplikationen und neurokognitive Auswirkungen.
Im Rahmen der HIT-Netzwerkaktivität werden zum einen die Spätfolgen bei Betroffenen systematisch erfasst und zum anderen im Rahmen der Therapieoptimierungsstudien risikoadaptiert deeskalierende Maßnahmen vorgenommen, aber auch therapeutische Aktivitäten in Richtung Spätfolgen entwickelt (z. B. neurokognitives Training; [53]).
CNS tumors of childhood and adolescence – basics and current developments
As an entity CNS tumors are the most common solid tumors of childhood and adolescence. Up to 500 cases are reported to the centres of excellence of the German HIT network every year. Low-grade gliomas comprise up to 50% of all CNS neoplasms in that age range. The most common malignant CNS tumor is medulloblastoma. While diagnostic measures such as neuropathology and radiology as well as CSF cytology have been performed on the highest level in reference laboratories of the HIT-network for decades, newer methods such as a methylation classifier have only recently amended this armamentarium.
In certain risk groups conventional therapy such as surgery, radio- and chemotherapy (including high-dose chemotherapy) may cure the majority of affected children. However, innovative approaches are desperately needed in high-risk populations such as diffuse H3K27-mutated midline gliomas, embryonal tumors such as ATRT or ETMR, but also metastatic CNS tumors in infants and other very young children.
A special focus of CNS tumor care lies on the early recognition and adequate response to side effects of the disease itself, but also on therapeutically induced early and late sequelae.
Keywords: CNS tumors, HIT network, methylation classifier, targeted therapy, late effects
Die Autoren sind ärztliche Wissenschaftler des Behandlungsnetzwerks HIT der
GPOH (http://www.kinderkrebsinfo.de). Die Studien des HIT-Netzwerkes werden von der Deutschen Kinderkrebsstiftung unterstützt (http://www.kinderkrebsstiftung.de).
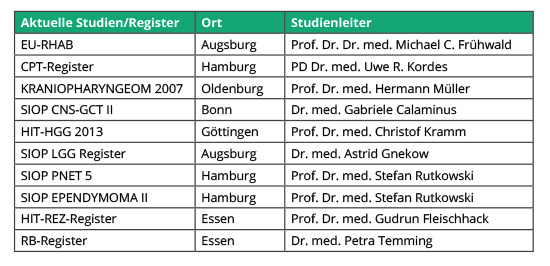
Die Daten aus folgenden Therapieoptimierungsstudien haben zu dieser Übersichtsarbeit beigetragen:
Autoren

