Vaskulitiden - Heterogener Krankheitskomplex
DOI: https://doi.org/10.47184/td.2024.02.08Vaskulitiden bilden eine sehr heterogene Gruppe im Rahmen der systemisch-rheumatologischen Grunderkrankungen. Ihnen gemeinsam ist eine Entzündung der Gefäße, wobei sämtliche Gefäße betroffen sein können. Die Symptome können je nach befallenen Gefäßen zum Teil stark variieren. Gefäßentzündungen können mit einem schweren Verlauf einhergehen, weshalb eine zügige Diagnose essenziell ist.
Schlüsselwörter: Riesenzellarteriitis, ANCA-assoziierte Vaskulitiden, Chapel Hill, ACR, EULAR, Therapie
Abkürzungsverzeichnis | |
|---|---|
AAV | ANCA-assoziierte Vaskulitiden |
CV | Kryoglobulinämische Vaskulitis |
EGPA | Eosinophile Granulomatose mit Polyangiitis, ehemals Churg-Strauss-Syndrom |
GCA | „giant-cell arteritis“ |
GPA | Granulomatose mit Polyangiitis, ehemals Morbus Wegener |
HUV | Hypokomplementärische Urtikariavaskulitis (Anti-C1q-Vaskulitis) |
KD | Kawasaki-Syndrom |
LVV | „large vessel vasculitis“ |
MPA | Mikroskopische Polyangiitis |
MVV | „middle vessel vasculitis“ |
PAN | Polyarteriitis nodosa |
PMR | Polymyalgia rheumatica |
RZA | Riesenzellarteriitis |
SVV | „small vessel vasculitis“ |
TAK | Takayasu-Arteriitis |
In der Rheumatologie gibt es unterschiedlichste Erkrankungen. Deren Einteilung erfolgt nach verschiedenen Kriterien: der Symptomatik, Labormarkern und Erstbeschreibern. Grob lassen sich fünf „Säulen eines Tempels“ der rheumatologischen Erkrankungen erkennen:
- Rheumatoide Arthritis
- Psoriasis-Arthritis (auch mit Befall der Wirbelsäule)
- Spondyl-Arthritis (ehemals Morbus Bechterew)
- Kollagenosen
- Vaskulitiden
Weitere Säulen stellen Kristallarthropathien und viral bedingte Entzündungen dar.
Unter Vaskulitiden versteht man eine heterogene Krankheitsgruppe, deren verbindendes Merkmal eine Entzündung der Gefäße darstellt. Ein Befall kann alle Arten von Gefäßen treffen: Arterien, Arteriolen, Kapillaren, Venolen und Venen. Im Jahr 2012 erfolgte eine Neueinteilung der Vaskulitiden nach der sogenannten Chapel Hill Consensus Classification (CHCC) [1] (Tab. 1a und 1b).
Tab. 1a: Einteilung von Vaskulitiden großer und mittelgroßer Gefäße [1, 2].
Nomenklatur gemäß Chapel Hill Consensus Conference (CHCC) 2012 mit englischen Abkürzungen | Klinische und diagnostische Besonderheiten |
|---|---|
Großgefäßvaskulitiden („large vessel vasculitis“; LVV), betreffen die Aorta und ihre Hauptäste sowie davon abgehend Arterien jeder Größe | |
Takayasu-Arteriitis (TAK) | Oft granulomatös, junge Patient:innen, weibliche Personen häufiger betroffen |
Riesenzellarteriitis (RZA) Giant-cell arteritis (GCA) (früher u. a. Morbus Horton) | Granulomatös, BSG („Sturzsenkung“), A. carotis mit Ästen (A. temporalis) und A. vertebralis; bei Pat. über 50 Jahre, männliche Personen häufiger betroffen, oft mit Polymyalgia rheumatica (PMR) assoziiert, Befall der Aorta (Aortitis) |
Vaskulitiden mittelgroßer Gefäße („middle vessel vasculitis“; MVV), häufig Aneurysmata und Stenosen | |
Polyarteriitis nodosa (PAN) | Nekrotisierend ohne Glumerulonephritis, keine antineutrophilen zytoplasmatischen Antikörper (ANCA) |
Kawasaki-Syndrom (KD) | Kutane Lymphknoten, Herz, bei Kindern |
Tab. 1b: Vaskulitiden kleiner Gefäße („small vessel vasculitis“; SVV) [1, 2].
Erkrankung | Pathophysiologie/Organbefall |
|---|---|
ANCA-assoziierte Vaskulitiden Pathologisches Muster: nekrotisierend mit geringer Immunkomplexablagerung (pauci immun) [3] | |
Granulomatose mit Polyangiitis (GPA) | Granulomatös, nekrotisierend, Lunge, Niere, Sinusitis, PR3(Proteinase 3)-ANCA (c-ANCA)-positiv |
Eosinophile Granulomatose mit Polyangiitis (EGPA) | Bluteosinophilie, Lunge, Niere, Sinusitis, z. T. ANCA-positiv (p3- und MPO-ANCA möglich) |
Mikroskopische Polyangiitis (MPA) | Nekrotisierend, Nierenbeteiligung |
Immunkomplexvaskulitiden Pathologisches Muster: Ablagerung von Immunkomplexen | |
Anti-GBM(glomeruläre Basalmembran)-Erkrankung (ehem. Goodpasture-Syndrom) | Lunge, Niere |
Kryoglobulinämische Vaskulitis (CV) (siehe auch Tab. 2) | Ablagerung von Kryoglobulinen in Haut, ZNS, Organen |
IgA-Vaskulitis | Häufigste Vaskulitis im Kindesalter: Glomerulonephritis, Gastrointestinaltrakt, Gelenke, Haut |
Hypokomplementärische Urtikariavaskulitis (HUV; Anti-C1q-Vaskulitis) | Glomerulonephritis, pulmonale Obstruktion, Arthritis, Auge |
Tab. 2: Spektrum der kryoglobulinämischen Vaskulitis-assoziierten Erkrankungen und Ursachen [4–6].
Erkrankung | Ursache |
|---|---|
Infektion | Hepatitis-C-Virus, Hepatitis-B-Virus, Humanes Immundefizienz-Virus (HIV), subakute bakterielle Endokarditis |
Kollagenose | Primäres Sjögren-Syndrom, systemischer Lupus erythematodes (SLE), Mischkollagenose („mixed connective tissue disease“; MCTD) |
Arthritis | Rheumatoide Arthritis |
Hämatologische Erkrankung | Lymphom, Plasmozytom, Leukämie |
Dabei wird zudem zwischen primären und sekundären Vaskulitiden unterschieden.
Epidemiologie
Etwa 200 von 1.000.000 Menschen leiden unter einer Vaskulitis – jährlich kommen 50 von 1.000.000 Menschen hinzu. Vaskulitiden in Gänze treten in allen Altersgruppen auf – allerdings gibt es Häufungen des Auftretens je nach Altersgruppe.
Zum Beispiel tritt eine Purpura Schönlein-Henoch vor allem bei Kindern nach Infekten auf; eine Riesenzellarteriitis befällt Personen, die älter als 50 Jahre sind.
Die häufigsten Vaskulitiden sind an erster Stelle die Riesenzellarteriitis (RZA), an zweiter Stelle die Kleingefäßvaskulitiden, die ANCA (antineutrophile zytoplasmatische Antikörper) aufweisen: die Granulomatose mit Polyangiitis (GPA; ehemals Morbus Wegener), die mikroskopische Polyangiitis (MPA) sowie die eosinophile Granulomatose mit Polyangiitis (EGPA; ehemals Churg-Strauss-Syndrom). Im Allgemeinen treten die Vaskulitiden im Norden häufiger auf (Nord-Süd-Gefälle) [6].
Symptomatik
Die Symptomatik der Vaskulitiden ist zwar unterschiedlich, doch gibt es einige allgemeine Symptome, die größtenteils gemeinsam zutreffen. Allen Vaskulitiden sind eine generelle Abgeschlagenheit sowie eine verschieden ausgeprägte B-Symptomatik gemein (Trias aus Fieber, Nachtschweiß und Gewichtsverlust).
Im Weiteren unterscheiden sich die Symptome nach Befall der Größe der Gefäße: Bei einer Kleingefäßvaskulitis können unter anderem eine Purpura, eine Episkleritis, eine Polyneuritis und ähnliche Erscheinungen beobachtet werden. Bei einem Befall der mittleren Gefäße werden unter anderem Thrombosen sowie Embolien mit Infarzierungen in den Nieren, im zentralen Nervensystem (ZNS) oder am Herzen beobachtet. Bei Großgefäßbefall ist, wenn die Gefäßwand entzündet ist, an eine Aortitis und eine Arteriitis temporalis zu denken: Die Betroffenen beschreiben eine deutliche Durchblutungsstörung einer Extremität oder zum Beispiel Symptome wie eine Kiefer- oder Zungenclaudicatio (Schmerzen beim Kauen) oder eine Amaurosis fugax, eine flüchtige Blindheit, auf einem Auge [7–10, 12–14].
Ausgewählte Erkrankungen
Wie bereits erwähnt, sind die Hauptvertreter der Vaskulitiden die Riesenzellarteriitis sowie die ANCA-assoziierten Vaskulitiden (AAV) GPA und MPA. Aus diesem Grund wird hier das Augenmerk auf diese beiden Gruppen gelenkt.
RZA
Die Riesenzellarteriitis tritt ab dem 50. Lebensjahr auf und geht mit einer unspezifischen B-Symptomatik einher. Im Rahmen dieser Erkrankung werden zwei Manifestationen unterschieden: ein zerebraler und ein extrazerebraler Befall. Unter dem zerebralen Befall versteht man die bisher als Morbus temporalis Horton bezeichnete Erkrankung; unter dem extrazerebralen Befall die „klassische“ Riesenzellarteriitis mit Befall der Aorta – einer Aortitis – sowie einem Befall der aus ihr abgehenden Gefäße, zum Beispiel Arteria carotis (Halsschlagader) oder die Arteria subclavia, die den Arm versorgt. Neue Kriterien für die RZA wurden 2022 veröffentlicht (Tab. 3).
Tab. 3: ACR/EULAR–Klassifikationskriterien 2022 für die Riesenzellarteriitis (RZA) [11]. Unter der Voraussetzung, dass der/die Betroffene ≥ 50 Jahre alt ist, wird wird nach einem Punktesystem vorgegangen. Die Klassifikation gilt als erfüllt, wenn sechs oder mehr Punkte erreicht werden. Die Sensitivität liegt bei 87 %, die Spezifität bei 95 %.
Klinische Kriterien | |
|---|---|
1. Morgensteifigkeit Schulter/Nacken | + 2 |
2. Plötzlicher Sehverlust | + 3 |
3. Kiefer- oder Zungenclaudicatio | + 2 |
4. Neuer temporaler Kopfschmerz | + 2 |
5. Pathologischer Temporalarterientastbefund | + 2 |
Untersuchungskriterien | |
1. BSG > 50 mm/h n.W., CRP-Wert > 10 mg/l | + 3 |
2. A. temporalis Biopsie + oder US Halo + | + 5 |
3. A. axillaris bds. Beteiligung | + 2 |
4. FDG-PET-Aktivität über gesamte Aorta | + 2 |
Ein spezieller Marker hierfür wurde bisher nicht in die gängigen Diagnose- und Klassifikationskriterien aufgenommen. In der Diagnostik spielt vor allem die Ultraschalluntersuchung eine große Rolle – hierin wird das sogenannte „Halo-Phänomen“ beschrieben (u. a. von Prof. Dr. W. Schmidt, Berlin [7–10], Abb. 1).
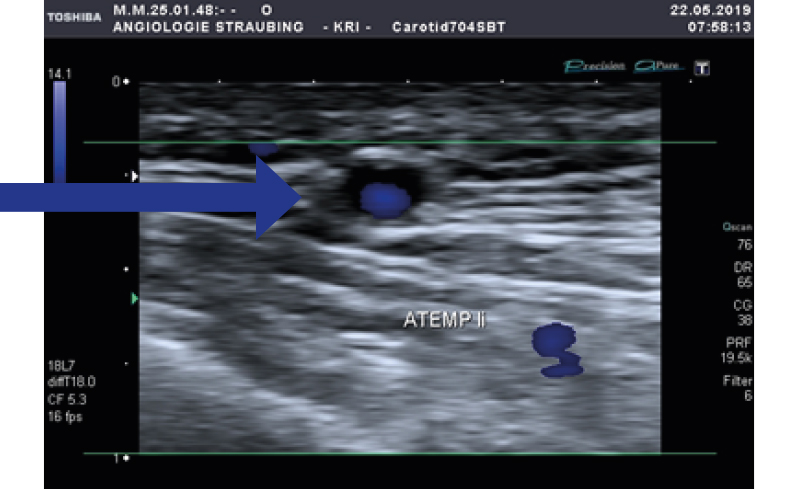
Abb. 1: Das Halo Sign (Bild: Autor).
AAV
Die Granulomatose mit Polyangiitis, Proteinase-3-ANCA-positiv, besteht aus einem Symptomkomplex mit chronischer, blutig-borkiger Sinusitis, einer Nierenbeteiligung (v. a. fokal-segmental nekrotisierende Glomerulonephritis) sowie einer Lungenbeteiligung mit pulmonalen Hämorrhagien bei pulmonaler Kapillaritis. Auch kann es zu einer entzündlich bedingten Trachealstenose kommen [1, 2, 12–17].
Die mikroskopische Polyangiitis, Myeloperoxidase-ANCA-positiv, ähnelt der eben genannten Symptomatik; der Unterschied besteht darin, dass bei der MPA – im Gegensatz zur GPA und auch zur EGPA – für gewöhnlich keine extravaskuläre, granulomatöse Komponente, die vor allem den Respirationstrakt betrifft, auffällt [1, 2].
Beiden genannten AAV liegen eine Autoantigen- und Erreger-getriggerte multifaktorielle Genese zugrunde [16, 20, 21]. Das Manifestationsalter liegt zwischen 45 und 70 Jahren, einen relevanten Geschlechterunterschied gibt es nicht [26]. Genetisch ist die GPA (PR3[Proteinase 3]-AAV) vor allem mit HLA(„human leukocyte antigen system“)-DP-Polymorphismen korreliert, wohingegen die MPA (MPO[Myeloperoxidase]-AAV) vor allem mit HLA-DQ-Polymorphismen assoziiert wird [16, 20, 23]. Die zum Toleranzverlust gegenüber der von Neutrophilen und Monozyten exprimierten PR3 und MPO führenden Faktoren sind bis heute nicht endgültig geklärt. Zyklische Schwankungen der Inzidenz einer Granulomatose mit Polyangiitis weisen auf einen möglichen Einfluss von Infektionen wie auch auf andere Umweltfaktoren hin [16, 20, 22]. Zudem ist die Assoziation der nasalen Staphylococcus-aureus-Besiedelung mit Rezidiven und Entzündungsaktivität im oberen Respirationstrakt bei der GPA bekannt – neuere Studien („next generation sequencing studies“) belegen eine Dysbiose des nasalen Mikrobioms, die dann mit der entzündlichen Aktivität korreliert [18–22].
Therapien
RZA
Die Therapie der Riesenzellarteriitis ist zunächst abhängig vom Schweregrad des entzündlichen Befalls und der durch die Betroffenen beschriebenen Symptomatik.
In der Therapie der Riesenzellarteriitis steht weiterhin eine hochdosierte Glukokortikoidtherapie an erster Stelle – in der Regel mit 1 mg Prednisolon pro Kilogramm Körpergewicht. Bei hochakuten Beschwerden – einem drohenden Visusverlust oder einer akuten Durchblutungsstörung einer Extremität – sollte die Therapie noch höher dosiert intravenös begonnen werden.
Zugelassen ist bisher neben der Glukokortikoidtherapie einzig eine Therapie mit Tocilizumab – einem IL-6-Rezeptor-Inhibitor. Eine Therapie mit Methotrexat (MTX) wird in Studien vielfach diskutiert und auch alltäglich durchgeführt, ist aber in dieser Indikationsstellung nicht zugelassen – zudem kann es unter laufender Therapie zu Rezidiven kommen [23].
AAV
Die Therapien der AAV – GPA und MPA – werden nach Chung et al. [24] wie folgt empfohlen: Zunächst wird zwischen einer „aktiven, schweren“ und einer „aktiven, nicht schweren“ Verlaufsform unterschieden. Bei der aktiven, schweren Verlaufsform wird ein Therapiebeginn bei beiden Erkrankungen mit Glukokortikoiden sowie Rituximab (RTX) vor Cyclophosphamid (CYC) empfohlen. Zum Remissionserhalt werden in dieser Reihenfolge anschließende Therapien empfohlen: 1. RTX, 2. MTX oder AZA (Azathioprin), 3. MMF (Mycophenolat-Mofetil) oder Leflunomid. Dies gilt für beide Erkrankungen.
Bei der aktiven, nicht schweren Verlaufsform werden nur bei der GPA Empfehlungen gegeben: Glukokortikoide in Kombination mit MTX vor RTX-, CYC-, AZA- oder MMF-Gabe. Ein Remissionserhalt erfolgt je nach Vortherapien mit MTX/AZA/MMF – nach Erhalt von RTX oder CYC wird auch RTX zum Remissionserhalt empfohlen.
Ein Plasmaaustauschverfahren wurde zuletzt bei GPA in der PEXIVAS-Studie relativiert [25]. In der 2021 publizierten ADVOCATE-Studie konnte die Wirksamkeit einer Kombination mit Avacopan – einem C5a-Rezeptor-Inhibitor – in Kombination mit RTX oder CYC in der Möglichkeit der schnelleren Glukokortikoidreduktion bei GPA/MPA gezeigt werden. Seit August 2022 ist diese Möglichkeit auch für beide Indikationen zugelassen [26, 27].
Zusammenfassung
Vaskulitiden bilden eine sehr heterogene Gruppe im Rahmen der systemisch-rheumatologischen Grunderkrankungen. Allerdings können sie mit einem schweren Verlauf einhergehen – die zügige Diagnose ist bei entsprechender Wachsamkeit zu postulieren.
Die Labordiagnostik kann mit spezifischen Autoantikörpern sowie mit Parametern zum Nachweis einer Entzündungsreaktion einen wichtigen Beitrag zur schnellen Diagnose leisten. Vaskulitis-assoziierte ANCA (VAA) sollten nach den aktualisierten Leitlinien bei der Fragestellung „AAV“ mithilfe spezifischer Immunoassays zum Nachweis von Anti-PR3- und Anti-MPO-Antikörpern durchgeführt werden. Als Bestätigungstest oder wenn möglich sogar parallel sollte der indirekte Immunfluoreszentest im Labor zur Verfügung stehen.
Unter entsprechender Medikation kann eine Remission erreicht werden.
