Die unterschätzte Kraft in der Immunologie
Mein Komplement!
Autoren: Prof. Dr. med. Rudolf Gruber und Dr. dr. med. Zsuzsanna Wolf
Defizienzen des Komplementsystems können durch Störungen der Immunfunktion zu einer erhöhten Infektanfälligkeit oder zu Autoimmunerkrankungen führen und angeborene Störungen der Komplementregulation zu Erkrankungen wie dem hereditären Angioödem und der paroxysmalen nächtlichen Hämoglobinurie. Es stehen bereits einige Tests zur Diagnostik von komplementassoziierten Erkrankungen zur Verfügung, eine Weiterentwicklung ist aber wünschenswert.
Schlüsselwörter: Complosom, membranattackierender Komplex, Mannose-/Mannan-bindendes Lektin, PNH-Therapie
Das Komplementsystem ist das „Schweizermesser“ des Immunsystems. Für die Pathophysiologie von Immunerkrankungen wurde es lange Zeit wenig beachtet. Die Aufmerksamkeit galt den T- und B-Zellen und den von den B-Zellen produzierten (Auto-)Antikörpern. Mit dem Einsatz potenter Komplementinhibitoren in der Therapie erkennt man mehr und mehr, dass eine fehlregulierte Aktivität des Komplementsystems bei entzündlichen und autoimmunen Erkrankungen eine wichtige Rolle spielt und in einigen Fällen die Grundlage der Erkrankung ist. Eine langfristige Behandlung des Komplementsystems kann aber auch zu unerwünschten Auswirkungen auf dieses homöostatische Gleichgewicht führen. Auch die Wechselwirkungen des Komplementsystems mit anderen Serum-Effektorsystemen wie dem Gerinnungs-, dem Renin-Angiotensin- und dem Kallikrein-Kinin-System werden zunehmend besser verstanden.
Complosom: Das intrazelluläre Komplementsystem
Relativ neue Forschungsarbeiten konnten zeigen, dass das Komplementsystem nicht nur im Blut aktiv ist, sondern auch eine wichtige Rolle bei der Entwicklung und Funktion von Geweben und Organen spielt. Beispielsweise kontrolliert das Complosom – wie die Gesamtheit der intrazellulären Elemente des Komplements genannt wird – die mitochondriale Aktivität, die Glykolyse und die oxidative Phosphorylierung. Des Weiteren sind Komplementfaktoren bei der Genregulation in angeborenen und adaptiven Immunzellen, aber auch zum Beispiel in Fibroblasten, Endothel- und Epithelzellen involviert.
Diese unerwartete Beteiligung des Komplementsystems an grundlegenden zellphysiologischen Aufgaben macht es zu einem neuartigen und zentralen Akteur bei der Kontrolle der Zellhomöostase. Diese Entdeckung sowie die Erkenntnis, dass man bei einer zunehmenden Zahl menschlicher Krankheiten Komplementstörungen als (Mit-)Ursache erkannt hat, haben das Interesse am Komplementsystem und seiner therapeutischen Beeinflussung neu entfacht. Störungen dieser Gewebeaktivität und von intrazellulären Komponenten des Komplementsystems sind mit einer Vielzahl von Krankheitsprozessen verbunden. So gibt es Hinweise, dass der intrazelluläre C5a-Rezeptor-1, der zum Beispiel auf Mitochondrien exprimiert ist, bei Kollagenosen und bei der Arteriosklerose dysreguliert ist und so zur Ausprägung des Krankheitsbildes beiträgt – oder dass sogar die Dysregulation eine ursächliche Rolle spielt [1]. Trotz guter Evidenz sind die Natur und die Funktionen der intrazellulären Komplementkomponenten in der Wissenschaft noch umstritten.
Komplement im Blut: (Patho-)Physiologie
Während das intrazelluläre Komplementsystem noch heftig diskutiert wird, gibt es vom extrazellulären Komplementsystem eine gut erforschte Grundidee der Funktionen. Wenn ein Krankheitserreger die Epithelbarrieren des Körpers überwunden hat, trifft er sehr früh auf eine Hauptkomponente der angeborenen Immunität, namentlich das Komplementsystem. Dabei handelt es sich um über 30 verschiedene lösliche und zellständige Proteine, die im Blut und anderen Körperflüssigkeiten sowie auf Zellen der Immunabwehr und weiteren Zellen vorkommen. Das Komplement wurde in den 1890er-Jahren von Jules Bordet als hitzeinstabile Komponente im normalen Plasma entdeckt, deren Aktivität die antibakterielle Aktivität von „Immunseren“ – also Antikörpern, wie wir jetzt wissen – „komplementiert“, das heißt ergänzt.
Die Nomenklatur der Komplementproteine ist leider etwas verwirrend. Die zuerst entdeckten Proteine gehören alle zum klassischen Reaktionsweg und sind durch den Buchstaben C und eine Zahl gekennzeichnet. Unglücklich ist dabei, dass sie in der Reihenfolge ihrer Erstbeschreibung nummeriert sind und nicht nach der Reaktionsreihenfolge. Beim klassischen Komplementweg ist das die Aktivierung von C1 → C4 → C2 → C3 und dann C5 bis C9 („membrane attack complex“; MAC). Neben dem klassischen Aktivierungsweg gibt es noch den alternativen und den Lektinweg. Ein vereinfachtes Schema der drei bekannten Komplementaktivierungswege ist in Abb. 1 dargestellt.
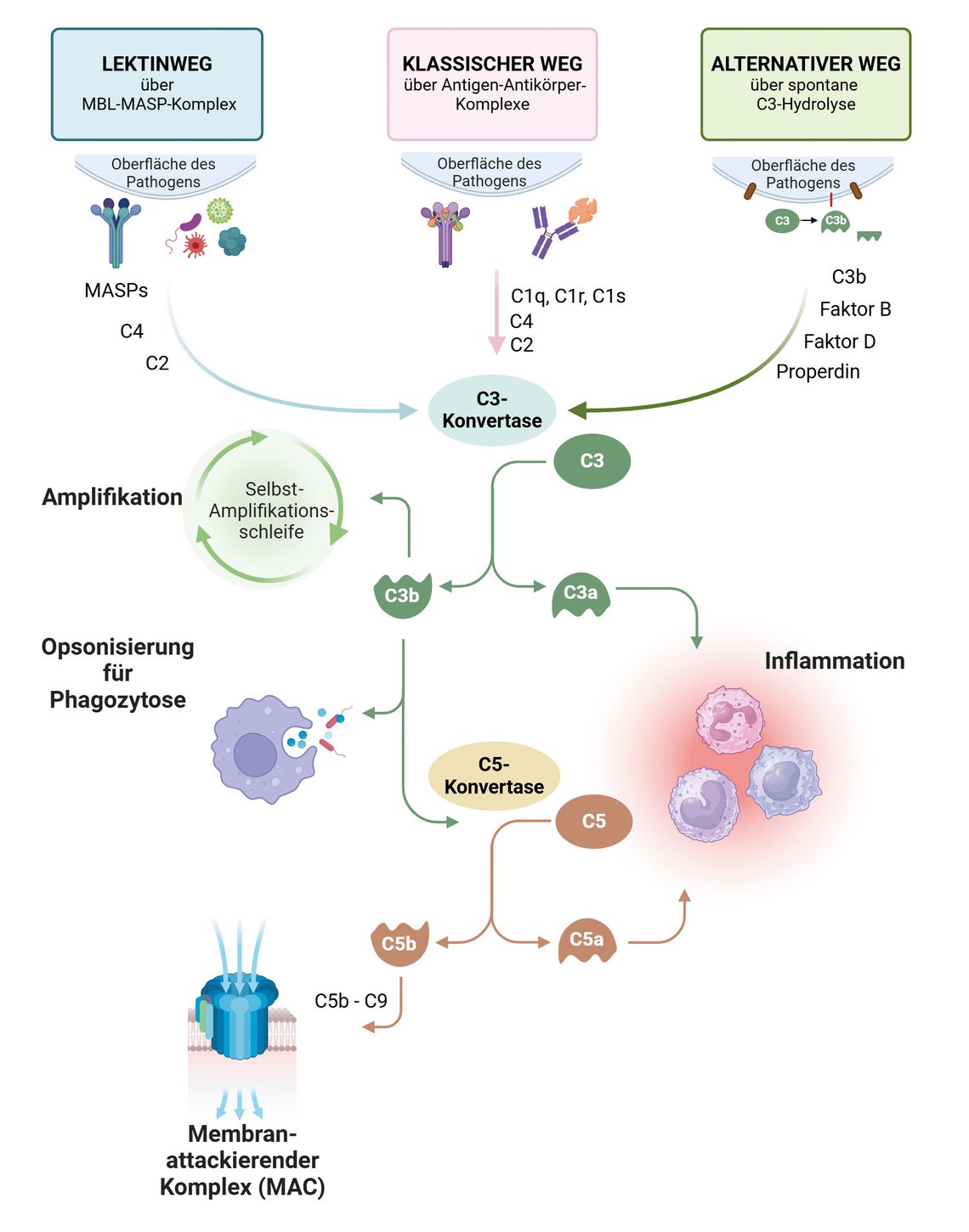
Abb. 1: Schematische Darstellung der drei Aktivierungswege der Komplementkaskade (Grafik: Zsuzsanna Wolf; die Abbildung wurde mit Biorender.com erstellt).
Wie man im Schema sehr schön sieht, ist die Initiierung der Aktivierung unterschiedlich, aber alle Wege treffen sich bei der C3-Konvertase und enden im membranattackierenden Komplex, also den Komponenten C5 bis C9. Der MAC kann Zellmembranen durchlöchern und so zur Lyse der attackierten Zelle führen. Diese Lyse von Zellen des Körpers einschließlich z. B. der roten Blutkörperchen, Bakterien und Parasiten ist ein Hauptwirkungsmechanismus des Komplementsystems. Auf dem Weg dorthin entstehen einige hochaktive Fragmente der Komplementfaktoren wie C3a und C5a, die für die Entzündungsreaktion eine wichtige Rolle spielen. Da es immer eine gewisse Grundaktivierung von Komplement im alternativen Weg gibt, gibt es auch ein System von Inhibitoren, die eine unkontrollierte Amplifikation verhindern sollen. Insbesondere die membranständigen Proteine CD55 („decay acceleration factor“; DAF) und CD59 sind hier zu erwähnen.
Klinik
Durch die Erforschung von Krankheiten bei Defizienzen von Komplementfaktoren konnten viele Funktionen entdeckt und genauer abgeklärt werden.
Komplementdefizienzen
Für nahezu alle Komplementfaktoren sind Mutationen beschrieben, die zu klinisch manifesten Erkrankungen führen können (Tab. 1). In der aktuellen Übersicht über alle bekannten Mutationen bei Immundefekten betreffen von 485 gelisteten Gendefekten 30 Mutationen das Komplementsystem. Hereditäre Komplementdefekte sind für ca. 5–10 % aller primären klinisch manifesten Immunmangelzustände ursächlich verantwortlich. Sie werden überwiegend autosomal rezessiv vererbt, weshalb heterozygote Träger klinisch meist unauffällig sind. Dabei gehört der genetisch determinierte Mangel des MBL (Mannose-/Mannan-bindendes Lektin) mit einer Prävalenz von bis zu 10 % zu den häufigsten Mutationen in Europa, die das Immunsystem betreffen. Ein klinisch sicher assoziierter Immundefekt ist eher selten, ein erhöhtes Risiko für Infektionen ist jedoch beschrieben [2, 3]. Zum Vergleich: Der häufigste klinisch manifeste Immundefekt mit einer Prävalenz von ca. 0,2 % ist der IgA-Mangel. Auch hier zeigen ca. 90 % der Betroffenen keine oder nur geringe Krankheitssymptome. Der symptomatische selektive IgA-Mangel ist überwiegend durch chronisch-rezidivierende, teils schwer verlaufende Atemwegsinfekte gekennzeichnet.
Bei C2-Defiziten oder auch Defekten in den Komponenten C1q, C1r, C1s oder C4 treten überwiegend Autoimmunerkrankungen auf, die unter anderem durch Immunkomplexe vermittelt sind. Ein kompletter kongenitaler C1q-Mangel ist der stärkste genetische Risikofaktor für die Entwicklung eines systemischen Lupus erythematodes (SLE). Eine Defizienz der Komponenten C3, C5–C8, Faktor H oder P führen zum Auftreten bakterieller Infektionen, oft durch bekapselte GRAM-negative Kokken (z. B. Neisserien). Bei Personen mit C5–C8-Defekten ist das Risiko einer Meningokokken-Erkrankung gegenüber der Normalbevölkerung 1.000- bis 10.000-fach erhöht.
Ein Fehlen von Faktor H führt zu einer unkontrollierten Aktivierung des alternativen Weges. An der Basalmembran der Nierenkörperchen und an der Bruchmembran des Auges kommt es zu C3-Ablagerungen. Das führt zu einer chronischen Nierenkrankheit (Membranoproliferative Glomerulonephritis Typ II), die mit Sehstörungen einhergehen kann. Die häufigere erworbene Form dieser Erkrankung wird durch einen Autoantikörper gegen den C3bBb-Komplex ausgelöst. Hier wird der Komplex stabilisiert und dadurch der alternative Weg aktiviert [4].
Interessant ist der Nachweis von kapillären C4d-Ablagerungen in den betroffenen Organen bei einer Transplantatabstoßung. C4d kann immunhistochemisch in einer Biopsie nachgewiesen werden. Diese Beobachtung hat wesentlich das Konzept der antikörpervermittelten Abstoßungsreaktion mitbegründet [5, 8].
Defekte Komplementregulatoren
Der weitaus häufigste, klinisch relevante Defekt eines Komplementregulators betrifft den C1-Esterase-Inhibitor (C1-INH) und führt zum Krankheitsbild des hereditären Angioödems (HAE). Durch die übermäßige Generierung von Anaphylatoxinen (C3a, C5a) mit Permeabilitätsstörungen der Gefäße kann es zu Schwellungen an Händen, Füßen, im Gesicht (Quincke-Ödem) und im Gastrointestinaltrakt (Koliken) kommen. Besonders gefährlich sind akute, anfallsweise auftretende Schwellungen von Larynx und Pharynx.
Neben der genetischen Form des C1-INH-Mangels (HAE Typ I; ca. 85 % der Fälle), die meist auch mit einem C4-Mangel einhergeht, gibt es weitere Formen: beispielsweise die genetisch bedingte Synthese eines dysfunktionellen Inhibitors (HAE Typ II), bei dem der C1-INH-Plasmaspiegel normal oder sogar erhöht, die Funktion aber reduziert ist. Als HAE Typ III bezeichnet man das Bradykinin-vermittelte Angioödem mit normalem C1-INH-Spiegel im Blut, aber z. B. Mutationen im FXII-Gen. Differentialdiagnostisch sind erworbene Angioödeme, die unter anderem durch ACE-Hemmer oder inhibierende Autoantikörper gegen den C1-INH ausgelöst werden, zu beachten und weiter die histaminvermittelten Haut- und Schleimhautschwellungen infolge von Allergien oder bei idiopathischer Urtikaria. Eine gesicherte Diagnose ist sehr wichtig, da sich die Therapie des HAE, das durch einen C1-INH-Mangel verursacht ist, grundlegend von der eines allergischen Angioödems oder gar einer Urtikaria unterscheidet. Ein Mangel kann durch die Substitution erfolgreich behandelt werden; Kortikosteroide und Antihistaminika zeigen dagegen nur bei allergischer Ursache oder der Urtikaria eine Wirkung.
Eine weitere wichtige Erkrankung, die durch den Mangel an Inhibitoren ausgelöst wird, ist die paroxysmale nächtliche Hämoglobinurie (PNH). Bei der PNH führt ein Defekt der sogenannten GPI-Anker (Glykosylphosphatidyl-Inositol-verankerte Oberflächenmoleküle) auf Blutzellen zum Verlust wichtiger Komplementinhibitoren. Es handelt sich um eine klonale Erkrankung der Hämatopoese, bei der pluripotente hämatopoetische Stammzellen aus dem Knochenmark einen charakteristischen Defekt aufweisen, nämlich eine Mutation des Gens für Phosphatidyl-Inositol-Glykan-A (PIGA).
Von diesem Defekt besonders betroffen sind Erythrozyten, da sie das Fehlen der Komplementregulatoren CD55 und CD59 nicht kompensieren können. Dies führt zu einer erhöhten Empfindlichkeit für die Lyse durch Komplement und in der Folge zur Hämoglobinurie. Erkrankungsschübe treten auf, wenn sich die pathologischen Klone stark vermehrt haben, dann durch einen Auslöser viele Erythrozyten auf einmal lysieren und das Hämoglobin über die Niere ausgeschieden wird. Daher der Name paroxysmale (anfallsartig) nächtliche (Betroffene bemerken den blutigen Urin meist beim ersten Urinieren am Morgen) Hämoglobinurie (Blut im Urin).
Kompliment ans Labor – die Komplementdiagnostik
Primäre Immundefekte aufgrund von Komplementdefekten sind selten. Die Aktivierung und der Verbrauch von Komplementfaktoren im Zusammenhang mit inflammatorischer Aktivität sind aber häufig, z. B. im Rahmen von Autoimmunerkrankungen und Infekten. Aktuell gibt es wohl eine Unterbewertung des Komplements, die dazu führt, dass zu selten an die Diagnostik der Komplementaktivität gedacht wird. Die geringe Anforderung der Diagnostik führt wiederum zu einer mangelnden Entwicklungsarbeit an neuen Testsystemen. Um die Komplexität der regulatorischen Netzwerke zu verstehen und Komplementdefizienzen angemessen zu berücksichtigen, sind umfassende diagnostische Tests erforderlich.
Wichtige Indikationen für eine differenzierte Komplementanalytik sind:
- Meningokokken-Meningitis,
- andere rezidivierende bakterielle Infektionen,
- Autoimmunerkrankungen,
- inflammatorische Erkrankungen der Niere und der Augen,
- Angioödem.
Die Möglichkeiten der Labordiagnostik sind vielfältig, aber aufgrund der Komplexität der Assays und der Seltenheit der Anforderung häufig nur in Speziallaboren etabliert. Funktionelle Globaltests können einen groben Überblick über die Gesamtfunktion geben. Hier gibt es hämolytische Tests wie CH50 (komplementvermittelte Hämolyse des klassischen Aktivierungsweges von 50 % der im Test eingesetzten, mit IgG beladenen Erythrozyten) und AH50 (alternativer Aktivierungsweg führt zur Hämolyse). Dies sind grundsätzlich einfache und kostengünstige Tests, doch sind sie durch den Einsatz von Erythrozyten nicht so gut standardisierbar wie Immunoassays und nur begrenzt haltbar. Alternativ gibt es Systeme mit Liposomen oder ELISAs. Ein ELISA kann differenziert Defizienzen aller drei Aktivierungswege funktionell in einem Ansatz nachweisen [6] (Tab. 1).
Tab. 1: Erkrankungen des Komplementsystems und dazugehörige Analytik (ohne genetische Untersuchungen) (modifiziert nach Kirschfink [4] sowie Kolde [9]).
Abkürzungen: aHUS = atypisches hämolytisch-urämisches Syndrom; AMD = altersabhängige Makuladegeneration; DAF = „decay accelerating factor“; FA = Funktionsassay; GN = Glomerulonephritis; IA = Immunoassay; NGS = Next Generation Sequencing; PCR = Polymerase-Kettenreaktion; PNH = paroxysmale nächtliche Hämoglobinurie; SLE = systemischer Lupus erythematodes; ST = Suchtest.
| Komponenten | Erkrankung | Testverfahren |
|---|---|---|
| Komplementfaktoren | ||
| C1q, C1r, C1s | Diverse Autoimmunerkrankungen, insbesondere SLE | ST (z. B. CH50), FA, IA |
| C2, C3 | Bakterielle Infektionen (GN), SLE | |
| C4 | Autoimmunerkrankungen, Angioödem | |
| C5, C6, C7, C8 | Rezidivierende Neisseria-Infektionen | |
| C9 | SLE | IA |
| Faktor B, Faktor D | Gehäufte Infektionen, aHUS | ST (z. B. AH50), FA, IA |
| Faktor H CFHR 1–5 | Infektionen mit Neisserien, aHUS, AMD, GN, SLE | FA, IA |
| Faktor I | Meningitis, andere Infekte, AMD, aHUS | ST (z. B. CH50), IA |
| Faktor P | Neisseria-Infektionen, Sepsis | ST (z. B. AH50), IA |
| MBL | Bakterielle Infektionen | ST, FA, IA |
| Inhibitoren (löslich) | ||
| C1-Inhibitor | Angioödem, Sepsis (Verbrauch) | FA, IA |
| Inhibitoren (Zellmembran) | ||
| DAF (CD55), CD59 | PNH | Durchflusszytometrie, Spezialtests |
| CR3 (CD11b/CD18) | Rezidivierende Hautinfektionen (Leukozytenadhäsionsdefekt; LAD) | Durchflusszytometrie |
| CD35/CR1 | SLE | Durchflusszytometrie |
| C5aR/CD88 | Chemotaxisdefekte | Durchflusszytometrie |
| Abbauprodukte | ||
| C3a, C3a-desArg, C5a | Akute Entzündung, Sepsis | IA |
| C4d | Nierenabstoßung | Immunhistochemie auf Nierenbiopsie |
| Autoantikörper | ||
| Anti-C1q | Hypokomplementämische Urtikaria, SLE | IA, FA |
| Anti-C1INH | Erworbenes Angioödem | IA, FA |
| Anti-fH | aHUS (HUS, MPGN) | IA, FA |
| C3-Nephritisfaktor | MPGN | IA, FA |
| Molekulare Genanalyse | ||
| Genetische Aufklärung/Charakterisierung von Komplementdefekten | PCR, NGS | |
| Detektion krankheitsassoziierter Mutationen und Polymorphismen | PCR, NGS | |
Ergibt sich hier ein auffälliger Befund, so kann die Bestimmung der Einzelkomponenten gut über Immunoassays abgedeckt werden. So hat sich die Standardanforderung „C3/C4“ in vielen Routinelaboren etabliert und ist auch auf Automaten standardisierbar. Mit „C3/C4“ bekommt man einen ersten Eindruck über einen zu hohen Verbrauch von Komplement, zum Beispiel im Rahmen von Autoimmunerkrankungen. Das reicht aber nicht aus, um die Vielfalt der Komplementaktivierung und mögliche Defekte abzudecken. Auch für die Bestimmung von Abbauprodukten der Komplementkaskade wie C3a oder C5a sind Immunoassays kommerziell erhältlich. Ihre Anwendung bleibt überwiegend Spezial- und Forschungslaboren vorbehalten. Dagegen hat sich die Bestimmung von wichtigen Autoantikörpern gegen Komplementfaktoren wie Anti-C1-Inhibitor oder C3-Nephritisfaktor gut in der Routine etabliert.
Eine klinisch sehr relevante, wenn auch seltene Anforderung ist die Bestimmung der membranständigen Komplementinhibitoren beispielsweise bei Verdacht auf PNH. Es gibt dazu Schnelltests, allerdings ist die Durchflusszytometrie die Methode der Wahl mit hoher Sensitivität und Spezifität, bei der der Nachweis des PNH-typischen GPI-Ankerdefekts auf Blutzellen erfolgt. Es sollten hier mindestens zwei unterschiedliche Reagenzien und zwei Zellreihen, zum Beispiel Granulozyten und Retikulozyten oder Monozyten, untersucht werden. Der Nachweis von GPI-verankerten Proteinen wie CD55/DAF und CD59 und/oder die Färbung mithilfe von FLAER („fluorescein-labeled proaerolysin“, ein direkt an das GPI-Ankermolekül bindendes Reagenz) kann standardisiert über kommerzielle Assays erfolgen. Nichtsdestotrotz ist die Analyse nicht trivial, und es ist viel Know-how beim Laborfachpersonal nötig, um die optimale Diagnostik zu gewährleisten.
Insbesondere beim Verdacht auf angeborene Komplementdefekte spielt die molekulare Genanalyse eine wesentliche Rolle.
Therapeutische Ansätze
Die Entwicklung neuer Therapien, wie zum Beispiel die C5-Inhibitoren Eculizumab und Ravulizumab, hat gezeigt, dass gezielte Interventionen am Komplementsystem für die Behandlung von verschiedenen Krankheiten sehr effektiv sein können.
Paroxysmale nächtliche Hämoglobinurie
Sehr gute Erfolge zeigte die Anwendung bei der PNH; sie hat sich hier trotz sehr hoher Kosten als Standardtherapie etabliert. Wie beschrieben führen somatische Mutationen im PIGA-Gen zu einem Defekt der Komplementinhibitoren CD55 und CD59. Das wiederum führt zu einer chronischen komplementvermittelten Hämolyse der betroffenen Erythrozyten. Der alternative Weg mit seinem konstanten Aktivierungszustand auf niedrigem Niveau trägt zur chronischen Natur der Hämolyse bei. Die intravaskuläre Hämolyse bedingt eine mittelschwere bis schwere Anämie. Eine extravaskuläre Hämolyse von Erythrozyten, die eine intravaskuläre Hämolyse überlebt haben, kann auch bei Personen auftreten, die mit Komplement-5-Inhibitoren behandelt werden. Die meisten Betroffenen sind auf wiederholte Transfusionen von Erythrozytenkonzentraten angewiesen. Durch die Behandlung mit Komplementinhibitoren haben nicht nur die Betroffenen einen klinischen Nutzen. Auch die Gesamtkosten der Patientenversorgung sind trotz hoher Therapiekosten geringer als bei nicht mit Inhibitoren behandelten Personen.
Atypisches hämolytisch-urämisches Syndrom
Eine weitere erfolgreiche Anwendung der C5-Inhibitoren ist das atypische hämolytisch-urämische Syndrom (aHUS). Das „typische HUS“ kann als Komplikation auf eine gastrointestinale Infektion mit Shigatoxin-produzierenden Escherichia coli (STEC) auftreten und wird daher auch STEC-HUS genannt. Hier konnte bisher keine eindeutige Wirkung von C5-Inhibitoren gezeigt werden, und es gibt bisher auch keine Zulassung. Das aHUS, auch komplementvermitteltes HUS, ist hauptsächlich mit Mutationen oder Autoantikörpern assoziiert, die zu einer dysregulierten Komplementaktivierung führen. Diese sehr seltene Form der thrombotischen Mikroangiopathie ist durch mikroangiopathische hämolytische Anämie, Thrombozytopenie und akutes Nierenversagen gekennzeichnet. Das Nierenversagen ist eine Folge einer Endothelzellschädigung der Nierengefäße. Ursache ist eine Dysregulation des alternativen Komplementweges an der Oberfläche der Endothelzellen, ausgelöst durch genetische oder erworbene Funktionsdefekte in Komplementregulatoren. Die Prognose der Betroffenen war vor der Einführung von Komplementinhibitoren schlecht; innerhalb von zwei Jahren nach der Diagnose entwickelten die meisten eine dialysepflichtige Nierenerkrankung. Die therapeutisch zur Verfügung stehenden Komplementinhibitoren können den fortschreitenden Verlust der Nierenfunktion verhindern. Dennoch bleiben erhebliche klinische und behandlungsbedingte Belastungen.
ANCA-assoziierte Vaskulitis
Eine bahnbrechende Entdeckung der vergangenen Jahre ist die Beschreibung der Rolle des Komplementsystems in der Pathogenese der ANCA-assoziierten Vaskulitis (AAV), insbesondere bei der pauci-immunen Glomerulonephritis bei AAV – pauci-immun („wenig immun“) genannt, da Immunkomplexablagerungen in den Gefäßwänden weitgehend fehlen. Heute weiß man, dass das eine unglückliche und irreführende Bezeichnung ist, da hier sehr wohl eine starke Beteiligung des Immunsystems in der Pathogenese vorliegt. Der 2022 zugelassene Wirkstoff Avacopan blockiert den Rezeptor für C5a – den aktivierten Komplementfaktor 5 – und damit dessen Wirkung, also zum Beispiel die Chemoattraktion von Neutrophilen. In Kombination mit einer Rituximab- oder Cyclophosphamid-basierten Induktionstherapie ist Avacopan für die Behandlung der schweren aktiven Granulomatose mit Polyangiitis (GPA) oder mikroskopische Polyangiitis (MPA) zugelassen [7]. Einen Überblick über die zugelassenen Medikamente mit Wirkung aufs Komplement einschließlich weiterer Krankheitsbilder ist in Tab. 2 dargestellt.
Tab. 2: Auswahl der aktuell zugelassenen Medikamente, die auf Komplementkomponenten wirken, sowie deren Indikationen (nach [10, 11]).
| Krankheitsbild | Medikamente | Bemerkungen |
|---|---|---|
| PNH (paroxysmale nächtliche Hämoglobinurie) | anti-C5 (Eculizumab, Ravulizumab) C3-Inhibitor (Pegcetacoplan) | Fehlen der Komplementregulatoren CD55 und CD59 |
| aHUS (atypisches hämolytisch-urämisches Syndrom) | anti-C5 (Eculizumab) | Genetische (seltener erworbene, z. B. Autoantikörper) Regulationsstörung im alternativen Weg des Komplementsystems; klinische Manifestation meist bei zusätzlichem exogenem Faktor, z. B. einer Infektion |
| Atypische PNH/hämolytisches Syndrom | anti-C5 (Eculizumab, Ravulizumab) | Mutationen der Komplementregulatoren CD55, CD59 oder anderer Faktoren; Autoantikörper |
| Refraktäre generalisierte Myasthenia gravis | anti-C5 (Eculizumab, Ravulizumab) | Bei Patient:innen ab 6 Jahren, die Acetylcholinrezeptor (AChR)-Antikörper-positiv sind |
| Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD) | anti-C5 (Eculizumab) | Bei Patient:innen, die positiv für Anti-Aquaporin-4 (AQP4)-Antikörper sind |
| ANCA-assoziierte Vaskulitis (schwere aktive Granulomatose mit Polyangiitis [GPA] oder mikroskopische Polyangiitis [MPA]) | C5aR1-Antagonist (Avacopan) | In Kombination mit einer Rituximab- oder Cyclophosphamid-basierten Induktionstherapie |
| Geografische Atrophie | C3-Inhibitor (Pegcetacoplan) | Spätstadium der trockenen altersbedingten Makuladegeneration (AMD) |
| Kälteagglutinin-Krankheit (Cold Agglutinin Disease; CAD) | anti-C1s (Sutimlimab) | Hämolytische Anämie bei erwachsenen Patient:innen CAD |
| Schwerer COVID-19-Verlauf | anti-C5a (Vilobelimab, FDA US-Notfallzulassung) | Innerhalb von 48 Stunden nach Beginn einer mechanisch-invasiven Beatmung (IMV) oder einer extrakorporalen Membranoxygenierung (ECMO) |
Ausblick
Der große klinische Erfolg der ersten Komplementinhibitoren führte zu weiteren Fortschritten bei der Entwicklung neuer Therapeutika, die aber bisher nicht aus der Nische der Anwendungen für Orphan Diseases (seltene Erkrankungen) herausgekommen sind. Die Verwendung der aktuell zugelassenen Inhibitoren, die alle auf die klassischen/Lektin- und terminalen Wege abzielen, ist mit Einschränkungen verbunden. So bleiben zum Beispiel einige Betroffene mit PNH anämisch und benötigen trotz der Behandlung mit einem C5- oder C3-Inhibitor weiterhin Bluttransfusionen. Eine weitere wichtige Nebenwirkung dieser Wirkstoffe ist das erhöhte Infektionsrisiko durch die Blockierung des terminalen Komplementweges, der eine Schlüsselrolle bei der Immunüberwachung spielt. Lebensbedrohliche Meningokokken-Infektionen und Infektionen durch bekapselte Bakterien, z. B. Streptococcus pneumoniae, Neisseria meningitidis und Haemophilus influenzae Typ B, sind möglich. Betroffene sollten daher einen Meningokokken-Impfstoff mindestens zwei Wochen vor Beginn der Therapie erhalten.
Neu entwickelte Wirkstoffe, die auf den alternativen Weg des Komplementsystems abzielen, bieten verbesserte Behandlungsmöglichkeiten sowohl für seltene Krankheiten wie die PNH und komplementvermittelte Nierenerkrankungen als auch für häufigere Leiden wie die altersbedingte Makuladegeneration (AMD). Präklinische Daten weisen darauf hin, dass einige dieser Wirkstoffe hochselektiv für den alternativen Weg sind und den klassischen Weg wie auch den Lektinweg intakt lassen, was das Infektionsrisiko im Vergleich zu C3- und terminalen Komplementinhibitoren senken könnte. Damit ist zu hoffen und zu erwarten, dass auch die Komplementdiagnostik weiterentwickelt wird. Selektive Inhibitoren erfordern eine spezifische Abklärung der verursachenden Komplementfaktoren. Auch für das Monitoring der Therapie könnte die Bestimmung der Komplementaktivität eine höhere Bedeutung bekommen.
Autoren


PDF Download

Sie können sich zum Lesen den Artikel auch als PDF herunterladen und anschließend den Fragebogen online ausfüllen.
Sie haben den Artikel gelesen?
Dann füllen Sie jetzt den Fragebogen aus. Sie erhalten 2 CME-Punkte, wenn Sie mindestens 7 von 10 Fragen richtig beantworten.
Jetzt Fragebogen ausfüllen