Diagnostik und Therapie der schweren kombinierten Immun-defekte im 21. Jahrhundert
Schwere kombinierte Immundefekte (SCID) sind eine heterogene Gruppe angeborener Erkrankungen der spezifischen zellulären und humoralen Immunität, die einer oftmals individuell angepassten Therapie bedürfen. Die therapeutischen Optionen haben sich in den vergangenen Jahren rasant vervielfältigt (Gentherapie, neue Methoden der haploidentischen HSZT, größere Anzahl registrierter Fremdspender). Auch diagnostisch wird mit dem Neugeborenenscreening eine neue Chance eröffnet, SCID-Patienten vor einer Beeinträchtigung durch schwerste Infektionen eine kurative Option anzubieten.
Schlüsselwörter: SCID, Immunphänotypen, Genetik, Stammzelltransplantation, Neugeborenenscreening
Einleitung
Schwere kombinierte Immundefekte (SCID für „Severe Combined Immunodeficiency“) sind eine Gruppe angeborener Immundefekte, die durch eine schwere Beeinträchtigung der zellulären und humoralen spezifischen Immunfunktionen definiert ist. Klinisch manifestiert sich die Erkrankung bereits im Säuglingsalter mit lebensbedrohlichen Infektionen. Kinder mit SCID versterben ohne Behandlung in der Regel vor dem zweiten Lebensjahr. Der Übergang von einem „schweren kombinierten Immundefekt“ (SCID) zu einem „kombinierten Immundefekt“ (CID) ist fließend. Die Zuordnung ist auch unter der Berücksichtigung klinischer, immunphänotypischer und genetischer Befunde in den ersten Lebensmonaten nicht für alle Patienten eindeutig möglich.
Klinische Präsentation
Infektionen
Die klinischen Manifestationen der Patienten mit SCID sind dominiert durch rezidivierende, ungewöhnlich schwere und somit atypische Verläufe von (opportunistischen) Infektionen. Beginn dieser Infektionen ist meist in den ersten Lebensmonaten. Betroffen sind häufig die Atemwege und der Gastrointestinaltrakt [1]. Typische Erreger sind Pneumocystis jirovecii, Adenovirus, Cytomegalievirus (CMV) oder das Respiratorische Synzytial-Virus (RSV). Eine Sonderrolle spielen Infektionen durch Lebendimpfungen: Kinder mit SCID fallen nach der seit 2013 von der STIKO empfohlenen Rotavirus-Lebendimpfung durch eine prolongierte Diarrhö mit Nachweis des Impfvirus auf. Dies ist ein klinisches Warnsignal, dem unbedingt nachgegangen werden sollte [2]. Auch andere Lebendimpfungen wie BCG oder die Polioschluckimpfung, welche in anderen Ländern regelhaft durchgeführt werden, können bei diesen Kindern zu lebensbedrohlichen Komplikationen führen.
Eine Sonderstellung nimmt die Retikuläre Dysgenesie ein: Neben dem Fehlen der Lymphozyten besteht eine Agranulozytose, sodass diese Kinder meist schon in den ersten Lebenstagen mit schweren bakteriellen Infektionen auffallen [3].
Maternofetale Transfusion
Maternale T-Zellen, die intrauterin an den Feten übertragen werden, können bei Fehlen der spezifischen zellulären Immunität nicht abgestoßen werden und persistieren im kindlichen Organismus. Hier treffen sie auf Alloantigene und können ein Bild verursachen, das klinisch dem einer Graft-versus-Host-Disease (GvHD) entspricht. Wie bei dieser können neben der Haut (Erythem) auch Darm (Enteritis) und Leber (cholestatische Hepatitis) beteiligt sein [4]. Diese Zeichen müssen sich nicht von Geburt an manifestieren und können sich auch erst in den ersten Lebensmonaten entwickeln. Die Differenzialdiagnose der Hauterscheinungen zu einem atopischen Ekzem oder Erythrodermien mit unterschiedlicher Ätiologie ist nicht immer einfach. Wichtig ist, bei diesen frühkindlichen Manifestationen an einen SCID zu denken und weitere diagnostische Schritte (s. unten) einzuleiten [4].
Als Ausdruck einer oligoklonalen peripheren Proliferation maternaler T-Zellen dominieren in der Immunphänotypisierung Gedächtnis-Zellen mit einem häufig eingeschränkten TCR-Repertoire [4]. Die maternalen T-Zellen können die Diagnostik erschweren: Expandierte maternale Zellen können zu normalen oder sogar erhöhten T-Zell-Zahlen im peripheren Blut führen, die HLA-Typisierung und genetische Diagnostik aus peripherem Blut kann durch die zusätzlichen mütterlichen Allele kompliziert werden. Bei Nachweis oder Verdacht auf eine MFT sollten genetische Untersuchungen und HLA-Typisierung nicht aus Vollblut oder Lymphozyten, sondern z. B. aus Granulozyten des peripheren Blutes durchgeführt werden.
Autoimmunität
In seltenen Fällen können autologe T-Zellen den klinischen Phänotyp eines SCID-Patienten wesentlich beeinflussen und die Diagnose erschweren. Auch kann eine ausgeprägte Leukozytose bestehen und den primären Verdacht in die Richtung einer leukämischen Erkrankung lenken. Der Nachweis autologer T-Zellen gelingt insbesondere bei Patienten mit hypomorphen Mutationen in Genen, die bei Nullmutationen einen „klassischen“ SCID-Phänotyp verursachen. Initial wurde dies bei Mutationen in den Recombinase Activating Genen (RAG1 oder RAG2) beobachtet, ist aber auch bei anderen Gendefekten beschrieben [5, 6]. Durch die Restfunktion der durch die hypomorphe Mutation veränderten Proteine, können einige T-Zellen ausreifen, treffen in ihrer Entwicklung jedoch auf eine insuffizient ausgebildete Thymusarchitektur, die keine zuverlässige Negativselektion autoreaktiver T-Zellen erlaubt. Diese wenigen autologen T-Zellen können bei Kontakt zu ihren Antigenen aktiviert werden und oligoklonal expandieren. Durch diese Autoimmunreaktionen können z. T. schwerste Organstörungen verursacht werden.
Diese besondere Manifestation des SCID durch Expansion autologer T-Zellen wird nach ihrem Erstbeschreiber als Omenn-Syndrom (OS) bezeichnet [7]. Patienten mit OS fallen durch Neurodermitis-ähnliche Exantheme, Alopezie, Kolitis bzw. Hepatitis ohne Erregernachweis auf, sowie durch Lymphadenopathien und Splenomegalie. Im Blutbild finden sich häufig eine Eosinophilie und im Serum z. T. deutlich erhöhte Spiegel für IgE. T-zelluläre Infiltrate mit eingeschränktem T-Zellrezeptor-Repertoire können sich in unterschiedlichen Geweben befinden, nicht unähnlich der GvH-Manifestation bei MFT (s. oben). Patienten jenseits des Säuglingsalters mit hypomorphen RAG-Defekten können durch Granulome an Haut, Schleimhaut und inneren Organen auffallen. Die histologisch epitheloidzelligen Granulome weisen T-zelluläre Infiltrate auf und dürfen nicht mit leukämischen Infiltraten verwechselt werden. Sie manifestieren sich z. B. an der Haut sowohl als papulöse als auch ulzerierende Effloreszenzen [8, 9]. Aus manchen dieser Granulome konnte Rötelnvirus isoliert werden, sodass ein infektiologischer Trigger postuliert wird [10].
Nicht-immunologische Manifestationen
Patienten mit SCID können neben der Infektneigung und Inflammationsreaktion durch maternale oder autologe T-Zellen weitere Auffälligkeiten zeigen. So kann ein Mikrozephalus Hinweis auf eine SCID-Entität mit Radiosensitivität sein: Mutationen in den Genen DNA-
Ligase IV, Cernunnos/XLF oder PKcs kodieren für Enzyme im DNA-Reparatur-Weg. Patienten mit radiosensitivem SCID haben ein erhöhtes Risiko für die Entwicklung von Tumoren und sollten Röntgen-, UV-Strahlung und bestimmte Chemotherapeutika (Alkylanzien) meiden. Veränderungen im DCLRE1C/Artemis-Gen gehen ohne Mikrozephalus einher, jedoch ebenfalls mit erhöhter Strahlensensitivität. Die retikuläre Dysgenesie oder Adenylatkinase (AK2)-Defizienz, welcher sowohl zu einer Agranulozytose als auch einem SCID führt, tritt in Kombination mit einer beidseitigen Innenohrtaubheit auf. Patienten mit Adenosin-Deaminase (ADA)-Mangel leiden an neurologischen Auffälligkeiten, welche auch nach Stammzelltransplantation persistieren.
Diagnostik
Immunphänotypisierung
Bei SCID liegt häufig – aber nicht immer – eine Lymphopenie vor, welche im Differenzialblutbild zu erkennen ist. Differenzialdiagnostisch sollte formell eine HIV-Infektion ausgeschlossen werden. Auch bei positiver Familienanamnese oder verdächtiger Klinik für SCID, sollte auch bei normalen Lymphozytenzahlen umgehend eine Untersuchung der Subpopulationen erfolgen. Definitionsgemäß fehlen beim klassischen SCID die T-Zellen (Ausnahmen sind MFT und ein Omenn-Syndrom mit autologen T-Zellen s. o.), sodass auch SCID-Patienten mit vorhandenen und primär unbeeinträchtigten B-Zellen keine Immunglobuline produzieren und ein „kombinierter“ Defekt für T- und B-Zellfunktionen vorliegt. In den ersten Lebensmonaten persistieren mütterliche Immunglobuline im kindlichen Organismus und sollten bei der Diagnostik berücksichtigt werden.
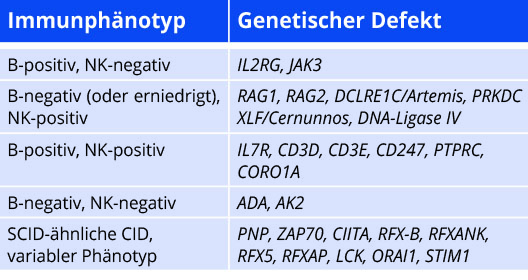
Die Phänotypisierung ermöglicht die Einteilung in sogenannte B-positive bzw. B-negative (sowie NK-positive und NK-negative) SCID-Varianten. Diese kann bereits einen ersten Hinweis auf den genetischen Subtyp geben.
Genetik
In Nordamerika und Europa verteilt sich die genetische Diagnose bei SCID etwa hälftig auf die X-chromosomal (verursacht durch hemizygote Mutationen in IL2RG) und die autosomal rezessiv vererbten Formen [11, 12]. Die Gendefekte variieren mit der ethnischen Zusammensetzung der Bevölkerung: RAG-Defekte sind in Nordamerika deutlich seltener als in europäischen Ländern. B-positive SCID-Varianten sind verursacht durch Defekte im IL2RG-Gen (kodiert für CD132/gemeinsame gamma-Kette mehrerer Zytokinrezeptoren) oder JAK3-Defekte. Hingegen führen einige Defekte in Genen, welche für Proteine der V(D)J-Rekombination kodieren (RAG1 und 2, DCLRE1C/Artemis), zu B-negativem SCID-Phänotyp. B-negative SCID-Varianten ohne NK-Zellen können Hinweis auf einen Mangel an ADA- oder AK2-Defekt (letzterer mit obligater Innenohrschwerhörigkeit) sein. Seltener werden Patienten mit B-positivem, NK-positivem SCID diagnostiziert, für welchen folgende Gendefekte ursächlich sein können: IL7RA, CD3D, CD3E, CD247, PTPRC/CD45 und CORO1A [13]. Hypomorphe Mutationen in DNA-Ligase IV, XLF/Cernunnos und PKc-Defekte (kodierend für Proteine der V(D)J-Rekombination) werden zwar unter der Gruppe der B-Zell-negativen SCID-Entitäten geführt[14]; da jedoch Mutationen mit Restfunktion vorliegen, ist ein breites Spektrum immunphänotypischer (positiver Nachweis von B-Zellen) und klinischer Manifestationen beschrieben[15].
Auch weitere Gendefekte wie ZAP70 oder MHC-Klasse-II-Defekte werden in der aktuellen PID-Klassifikation [16] meist als kombinierte Immundefekte aufgeführt, können jedoch ebenfalls zu diesem schweren Krankheitsbild führen und ein ähnliches Management erforderlich machen wie bei Patienten mit klassischem SCID.
Die Zuordnung zu einem Gendefekt ist nicht nur von akademischem Interesse, sondern kann von therapeutischer Relevanz sein, z. B. bei der Überlegung, ob eine Chemokonditionierung vor Stammzelltransplantation empfehlenswert ist und aus welchen Therapeutika sich diese zusammensetzen sollte. Letztlich ist auch die Differenzialdiagnose des DiGeorge-Syndroms (DGS) zu berücksichtigen, da ein Patient mit immunologisch komplettem DGS (keine T-Zellen im peripheren Blut) eine klinische Präsentation wie ein Patient mit SCID zeigen kann. Patienten mit DGS zeigen häufig zusätzliche kardiale und endokrinologische Auffälligkeiten (conotrunkale Herzvitien, Hypocalcämie durch Hypoparathyreoidismus) und faziale Dysmorphien. Eine gezielte Untersuchung dieser zusätzlichen klinischen Zeichen und ggf. ein genetischer Ausschluss sind anzustreben (z. B. Mikrodeletionssyndrom 22q11).
Für die Therapie der Patienten hätte die Diagnose eines DGS weitreichende Konsequenzen, eine Stammzelltransplantation ist hier nicht der primär kurative Ansatz, eher eine Thymusgewebs-Transplantation, die jedoch nur an wenigen Zentren weltweit durchgeführt wird.
Neugeborenenscreening
Eine Frühdiagnose des SCID ist bei Neugeborenen möglich, da diese Patienten entweder keine oder zu wenige thymusgereifte T-Zellen haben. Ein Surrogatmarker für diese naiven T-Zellen sind die T-cell receptor excision circles (TREC), DNA-Stücke, welche bei der Rekombination der TCR α-Kette des T-Zellrezeptors anfallen und per real-time Polymerasekettenreaktion quantitativ messbar sind. SCID-Patienten mit T-Zellen aufgrund maternofetaler Transfusion haben zwar T-Zellen, aber niedrige TREC-Werte und werden ebenfalls erkannt. Bei Patienten mit hypomorphen Gendefekten mit Restfunktion oder kombinierten Immundefekten, z. B. ZAP70 oder MHC-Klasse-II-Defizienz, können TREC-Werte noch in den Normbereich fallen, sodass das Neugeborenenscreening (NGS) die klassischen SCID-Varianten zwar detektiert, aber eine SCID-ähnliche Erkrankung oder andere angeborene Immundefekte nicht ausschließt. Umgekehrt werden durch das SCID-Screening auch einige Patienten mit Thymusdefekten (komplettes DGS), Patienten mit Ataxia teleangiectasia oder sekundären T-Zell-Lymphopenien erkannt. In Nordamerika wurde das SCID-Screening bereits vor einigen Jahren schrittweise eingeführt (erster USA-Staat war 2006 Wisconsin). Die Prognose von SCID-Patienten, welche im NGS auffallen, ist ausgezeichnet mit 95% Überleben, wenn sie frühzeitig und infektfrei einer kurativen Therapie (Stammzelltransplantation, bei ADA-Mangel auch Gentherapie) zugeführt werden [11, 17]. Die Prognose bei Kindern mit SCID, die nach schweren Infektionen transplantiert werden, ist vergleichsweise schlechter [18, 19]. In Deutschland ist die Aufnahme der TREC-Messung als Teil des freiwilligen NGS beantragt und wird demnächst angeboten werden können [20].
Therapie
Primäres Ziel der Therapie bei Patienten mit SCID ist die Etablierung eines T-Zell-Systems. Hierdurch werden (weitere) lebensbedrohliche Infektionen weitgehend vermieden. Die aktuell am häufigsten eingesetzte zelluläre Therapie mit diesem Ziel ist die hämatopoetische Stammzelltransplantation HSZT) [21]. Die gentherapeutische Manipulation autologer Stammzellen erfuhr einen schweren Rückschlag, da ein Großteil der Patienten, deren Stammzellen mit einem retroviralen Vektor manipuliert wurden, infolge der Therapie an einer lymphoblastischen Leukämie erkrankten. Aktuell werden in Studien andere Vektoren mit einem deutlich besseren Nebenwirkungsprofil bei Patienten mit Mutationen der IL2RG-Kette erprobt [21]. Für Patienten mit ADA-Mangel gibt es seit 2016 eine europäische Zulassung für die somatische Gentherapie, falls ein HLA-kompatibles Geschwisterkind als gesunder Stammzellspender nicht zur Verfügung steht [22].
Durch die schwere Defizienz der spezifischen Immunität bei Patienten mit SCID ergeben sich in der Transplantationsimmunologie einige Besonderheiten. Bei Verfügbarkeit eines HLA-identischen Familienspenders ist eine Transplantation ohne Konditionierung in vielen Fällen erfolgreich und kann sogar zu einem vollständigen Spenderchimärismus führen. Das Engraftment T-Zell-depletierter Transplantate von HLA-haploidentischen Spendern ist ohne Konditionierung jedoch erschwert. Die Abwägung der Intensität einer konditionierenden Chemotherapie vor HSZT ist bei Patienten somit eine individuelle Entscheidung und wird von Parametern wie dem klinischen Allgemeinzustand, Immunphänotyp, genetischer Entität, Infektionsmuster, Spenderverfügbarkeit, Transplantat-manipulation usw. beeinflusst [23].
Die häufigsten SCID-Entitäten zeichnen sich auch durch eine primäre Störung der Entwicklung oder Funktion der B-Zellen aus (z. B. IL2RG, JAK3, RAG1/2, DCLRE1C/Artemis), sodass die Rekonstitution der spezifischen humoralen Immunität vom Engraftment der Spender-B-Zellen abhängt. Findet dies nicht statt, sind langfristig – analog zur Agammaglobulinämie – Immunglobuline zu substitutieren. Eine Konditionierung verbessert die Wahrscheinlichkeit eines B-Zell-Engraftments, das mit dem Engraftment von Stammzellen korreliert. In der mittel- und langfristigen Nachsorge, haben Patienten nach Konditionierung auch einen höheren Anteil naiver, im Empfängerthymus gereifter T-Zellen [17].
