Von-Willebrand-Syndrom: Ein Syndrom – viele Gesichter
DOI: https://doi.org/10.47184/td.2022.04.02Das Von-Willebrand-Syndrom kann sowohl durch angeborene als auch durch erworbene quantitative oder qualitative Veränderungen des Von-Willebrand-Faktors verursacht werden. Neben Blutungsanamnese und Basis-Gerinnungsuntersuchungen werden bei der erweiterten Diagnostik auch das VWF-Antigen, die VWF-Aktivität und die Faktor-VIII-Aktivität bestimmt.
Schlüsselwörter: VWF:Ag, VWF:RCo, VWF:CB, VWF:GPIbM, VWF:GPIbR, FVIII:C
Beim Von-Willebrand-Syndrom (VWS) handelt es sich um die am häufigsten auftretende hämorrhagische Diathese in Deutschland. Die Prävalenz liegt beim Labor-Screening zwischen 0,8 und 1,3 %; Männer und Frauen sind gleichermaßen betroffen. Man unterscheidet eine angeborene und eine seltenere, erworbene Form, die durch multiple Grunderkrankungen (v. a. lymphoproliferative Erkrankungen, strukturelle Herzerkrankungen), Antikörper gegen den Von-Willebrand-Faktor (VWF), verminderte Synthese, erhöhte Proteolyse u. a. verursacht werden kann [1].
Der VWF wird in den Gefäßendothelzellen und Megakaryozten synthetisiert. Ein Teil davon zirkuliert frei im Blut. Das primäre Produkt besteht aus einem Prä-pro-VWF, einem Monomer, das komplexen biochemischen und strukturellen Veränderungen unterzogen wird. Das Resultat sind Multimere (Untereinheiten) gleicher Zusammensetzung. Diese sind in Abhängigkeit von der Anzahl der Monomere von unterschiedlicher Größe (500–20.000 Kd). Durch diese repetitive Zusammenlagerung von jeweils zwei identischen Bausteinen (Dimere) entstehen Moleküle mit bis zu 20 Untereinheiten, was den VWF zum größten löslichen Protein des menschlichen Organismus macht. Hochmolekulare Multimere sind wegen der höheren Anzahl an Bindungsstellen dabei die effektivste Form des VWF.
Die wesentlichen Aufgaben des Moleküls sind der Schutz des Gerinnungsfaktors VIII vor einer proteolytischen Degradation und die Rolle in der primären Hämostase. Hier agiert der VWF als Adhäsivfläche zwischen subendothelialen Strukturen und den Thrombozyten (GP Ib/V/IX) sowie zwischen den Blutplättchen untereinander (Abb. 1).
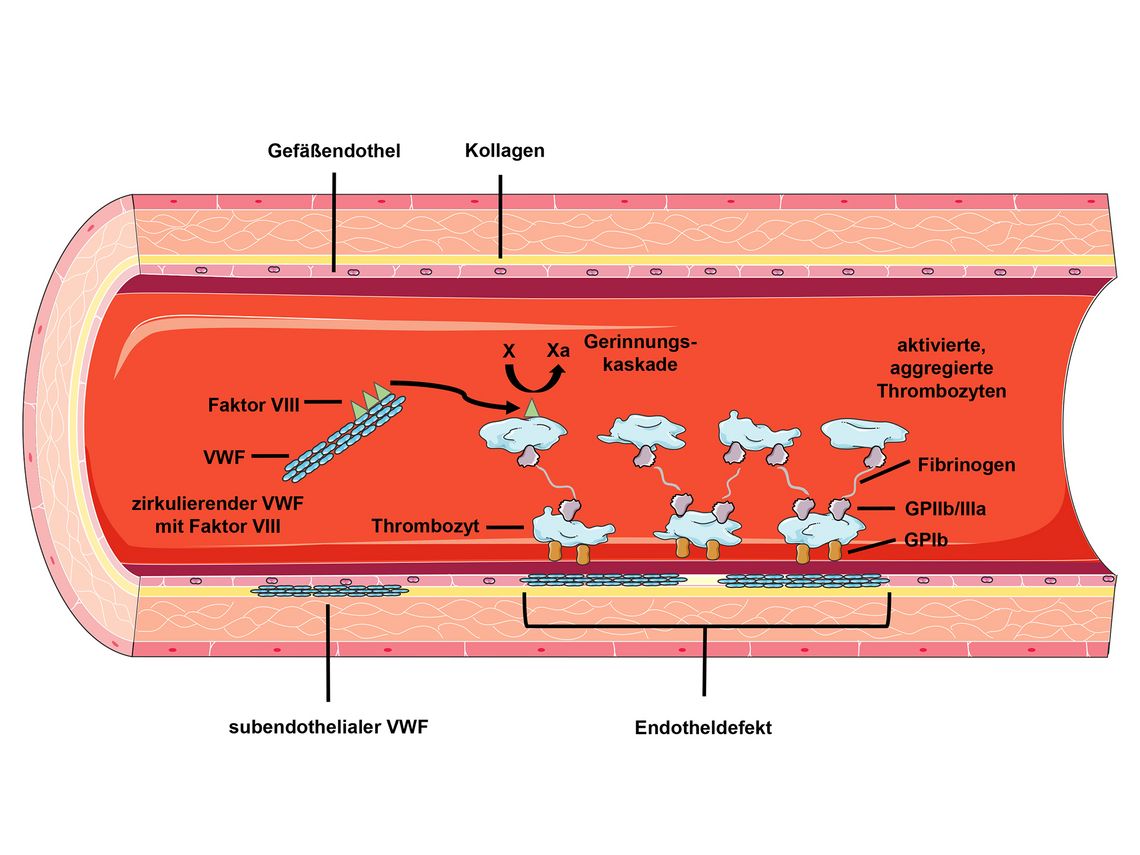
Abb. 1: Rolle des Von-Willebrand-Faktors in der Hämostase (Bild: Autor/Sabine Ramspott, Grafikbestandteile: smart.servier.com).
Einteilung
Die „International Society of Thrombosis and Haemostasis“ (ISTH) definiert die Erkrankung im Wesentlichen anhand der Konzentration des VWF und dessen Funktion [2]. Es werden dabei Typen klassifiziert. Die Typen 1 und 3 beschreiben quantitative, der Typ 2 (2A, 2B, 2M, 2N) qualitative Defekte. Bei Typ 1 ist der VWF leicht bis moderat vermindert, funktionell aber intakt. Mit ca. 70 % ist das Vererbungsmuster hier autosomal dominant. Der seltene Typ 3 ist mit einem schweren VWS assoziiert. Dieser Typ zeigt eine normale Struktur der Multimere; die Konzentration des VWF ist hingegen signifikant reduziert oder gleich null; infolgedessen ist auch der Faktor VIII vermindert. Das Vererbungsmuster ist hier autosomal rezessiv.
Bei den qualitativen Formen des VWS unterscheidet man vier wesentliche Unterformen: Bei Typ 2A kommt es zu einer Störung der Interaktion zwischen dem VWF und Thrombozyten durch den Verlust von Bindungsstellen bei Minderung der hochmolekularen Multimere. Hingegen ist bei Typ 2B die Affinität des VWF zu dem Thrombozytenantigen GPIb erhöht. Bei Typ 2M liegt ein Defekt der Interaktion zwischen Thrombozyten und dem VWF bei normalem Multimermuster vor. Der Typ 2N weist eine Mutation an der FVIII-Bindungsstelle auf, wodurch die Konzentration dieses Gerinnungsfaktors vermindert ist; die Struktur der Multimere ist bei diesem Subtyp jedoch unauffällig (Tab. 1).
Tab. 1: Einteilung des Von-Willebrand-Syndroms.
| Häufigkeit | Vererbung | Blutungsneigung |
|
|---|---|---|---|---|
Quantitative Defekte | ||||
Typ 1 (VWF vermindert) | ca. 58 % [3] | autosomal dominant | leicht bis moderat | Leichte bis moderate Minderung eines ansonsten qualitativ-strukturell unauffälligen VWF |
Typ 3 (VWF fehlt) | ca. 2 % [3] | autosomal rezessiv | schwer | Signifikante Reduktion eines ansonsten strukturell unauffälligen VWF (Multimere) |
Qualitative Defekte | ||||
Typ 2 (A, B, M, N) | ca. 40 % [3] | autosomal dominant | variabel |
|
Typ 2A |
|
|
| Reduzierte Plättchen-abhängige Funktion, Verminderung der hochmolekularen VWF-Multimere |
Typ 2B |
|
|
| Erhöhte Affinität für GPIb auf den Oberflächen der Thrombozyten, Verminderung der hochmolekularen VWF-Multimere |
Typ 2M |
|
|
| Reduzierte VWF-abhängige Adhäsion der Thrombozyten; funktioneller Defekt des VWF trotz Präsenz normaler, hochmolekularer Multimere |
Typ 2N |
|
|
| Normale Multimere; Faktor-VIII-Bindungskapazität reduziert |
Die gegenwärtige Klassifikation basiert vorwiegend auf einer Deskription des Phänotyps. Künftig werden zunehmend weitere Erkenntnisse biochemischer und molekularer Diagnostik Einzug finden, die eine Konkretisierung der Phänotyp/Genotyp-Korrelation nach sich ziehen sollen.
Klinische Symptome
Das VWS kann in jedem Alter symptomatisch werden. Eine Blutungsmanifestation (ca. 1 : 3.000 bis 1 : 10.000) ist dabei niedriger als die Prävalenz des VWS. Diese ist dabei sehr unterschiedlich ausgeprägt. Circa 70 % der Betroffenen haben eine milde Verlaufsform. Leitsymptom des klassischen VWS sind verlängerte Schleimhautblutungen (v. a. Epistaxis, Gingivablutung, bei Frauen eine verlängerte bzw. verstärkte Menstruation). Aufgrund der erniedrigten Aktivität des Faktors VIII bei Typ 2N gleicht das klinische Bild dem einer Hämophilie mit Weichteilblutungen, Blutungen nach Operationen oder Gelenkblutungen. Bei Typ 3 können sich beide Blutungstypen zeigen. Der Blutungstyp, die Blutungsfrequenz und der klinische Schweregrad sind ferner von anderen modulierenden Erkrankungen und Spätfolgen des VWS abhängig [3]. Hierzu zählen kardiale Erkrankungen, Teleangiektasien und Angiodysplasien, vermutlich durch eine Dysregulation angiogenetischer Mediatoren [4]. Die klinische Symptomatik ist interindividuell und kann sich in einem phasenhaften Verlauf präsentieren, zum Teil mit mehrjährigen symptomlosen Intervallen.
Labordiagnostik
Grundlage der Diagnostik des VWS ist eine Blutungsanamnese, die mit einer Familienanamnese assoziiert wird. Zur objektiven Erfassung der subjektiv beschriebenen Blutungssymptome stehen standardisierte Fragebögen zur Verfügung [5].
Die Heterogenität der Klinik und Beeinträchtigung des VWF spiegeln sich in der Laboranalytik in Parametern der primären und sekundären Hämostase wider. Zu den Basisuntersuchungen zählen die wenig standardisierte Blutungszeit nach Ivy, Globalparameter wie die aPTT (Faktor-VIII-Minderung), die jedoch nur bei ausgeprägten Formen des VWS pathologisch ist bzw. im Rahmen der Differentialdiagnostik genutzt wird (z. B. Hämophilie) und das Blutbild (v. a. Hb, Thrombozytenzahl). Für das Screening wird häufig auch die sogenannte In-vitro-Blutungszeit mittels Thrombozytenfunktiontest (z. B. PFA-100) eingesetzt, die eine hohe Sensitivität für bestimmte Typen des Von-Willebrand-Syndroms aufweist [6].
In der erweiterten Diagnostik werden das VWF-Antigen (VWF:Ag), die VWF-Aktivität (z. B. VWF:RCo, VWF: CB, VWF:GPIbM, VWF:GPIbR) und die Faktor-VIII-Aktivität (FVIII:C) bestimmt. Der Faktor VIII ist mit Ausnahme des Typs 2N nur mittelbar beeinträchtigt. Die Bestimmung des VWF:Ag ermöglicht die Differenzierung zwischen einem verminderten und dysfunktionellen VWF. Der Parameter ist bei ca. vier Fünfteln der Betroffenen mit VWS erniedrigt [7].
Die Analyse der VWF-Aktivität ist ein relevanter Parameter zur Erfassung der funktionellen Aktivität des VWF. Zu berücksichtigen sind die verkürzte Halbwertszeit des VWF bei Patient:innen mit der Blutgruppe 0 (bis zu 25 % niedrigere Konzentrationen des VWF), die Zunahme der Aktivität des VWF und des Faktors VIII im Rahmen akuter Phase-Reaktionen und die Altersabhängigkeit mit einem Anstieg ab dem 40. Lebensjahr. Eine Abhängigkeit vom Geschlecht besteht nicht.
Bei den VWF-Aktivitätstests existieren verschiedene Optionen: Für lange Zeit stellte die VWF:RCo-Methode den Goldstandard dar. Das Verfahren ist jedoch schwer zu standardisieren und erschwert hierdurch die Diagnose des Typ 2 (Ratio VWF:RCo/VWF:Ag). Möglicherweise wird hier künftig der Einsatz von plasmatischem oder rekombinantem GPIb in einem ELISA vermehrt zum Einsatz kommen [8]. Neben der Bindung an Thrombozyten kann bei Aktivitätstests auch die Bindung an Kollagen des verletzten Endothels (Bindungsstellen A1- und A3-Domänen) gemessen werden (VWF:CB) (Abb. 2, Tab. 2).
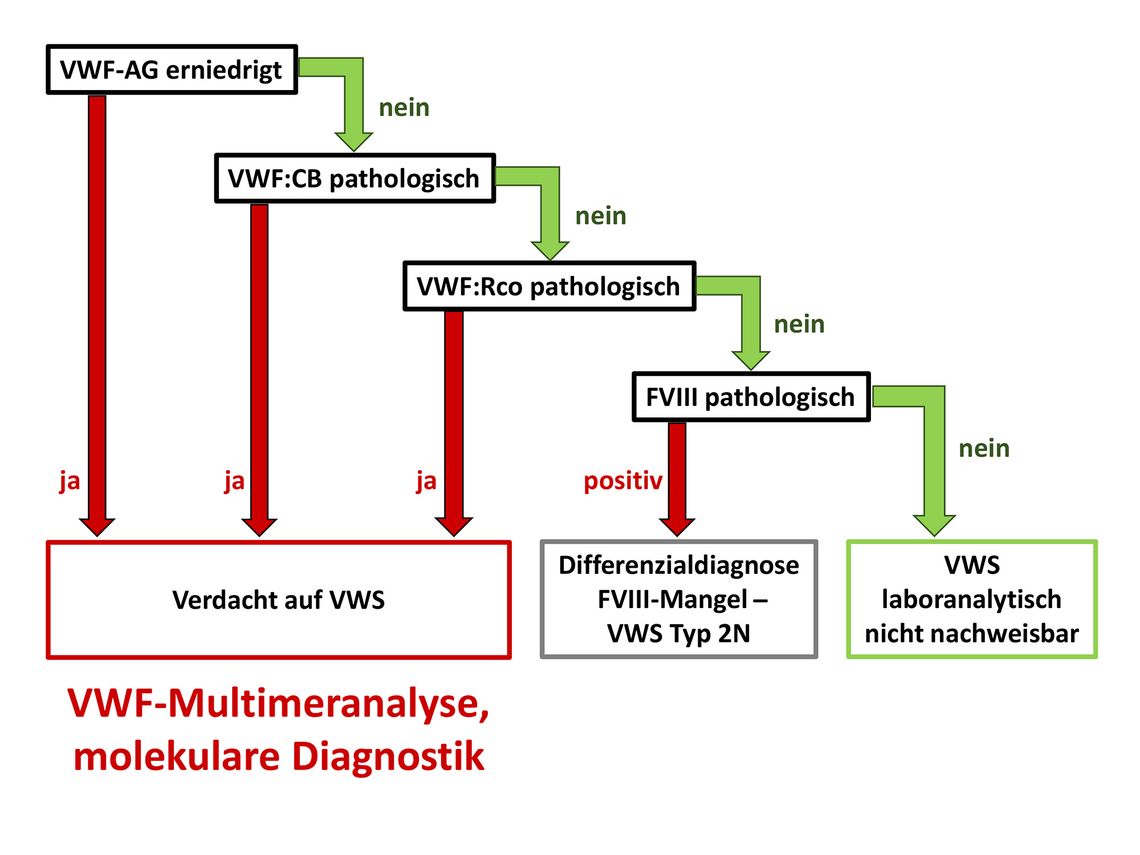
Abb. 2: Orientierendes Fließschema der Labordiagnostik des VWS (Grafik nach Autor).
Tab. 2: Laboranalytik und korrespondierende Klassifizierung des VWS (nach [11]).
Labortest | Hinweis auf | VWS Typ 1 | VWS Typ 2A | VWS Typ 2B | VWS Typ 2M | VWS Typ 2N | VWS Typ 3 |
|---|---|---|---|---|---|---|---|
PFA-100 (Verschlusszeit) | Primäre Hämostase, Thrombozytenfunktion | verlängert oder normal | verlängert, kein Verschluss | verlängert, kein Verschluss | verlängert, kein Verschluss | normal | verlängert, kein Verschluss |
FVIII:C (Faktor-VIII-Aktivität) | Interaktion FVIII mit VWF | niedrig oder normal | niedrig oder normal | niedrig oder normal | normal oder niedrig | niedrig | niedrig |
VWF:Ag | Konzentration des VWF | niedrig | niedrig oder normal | niedrig oder normal | normal oder niedrig | normal oder niedrig | sehr niedrig |
VWF:RCo | Funktion des VWF | niedrig | sehr niedrig | niedrig | niedrig | normal oder niedrig | sehr niedrig |
VWF:RCo / VWF:Ag Ratio | Verhältnis VWF-Aktivität zu VWF-Konzentration | normal | niedrig | niedrig | niedrig oder normal | normal | variabel |
RIPA | Interaktion VWF mit Plättchenrezeptor (GPIb) | reduziert oder normal | reduziert oder normal | gesteigert | reduziert oder normal | normal | keine Aggregation |
VWF-Multimeranalyse | Quantität, Größenverteilung und Konformitätsänderung des VWF-Moleküls | normales Muster, VWF reduziert | große und mittelgroße Multimere fehlen | große Multimere fehlen | normale Multimeren-verteilung | normal | keine Multimere vorhanden |
Bei der angeborenen Form können diverse Mutationen nachgewiesen werden. Diese führen zu einer veränderten Transkription des VWF und einer reduzierten Expression, zu Störungen des intrazellulären Dimer-Transports und gestörter Multimer-Bildung, zu einer erhöhten Spaltung des VWF durch ADAMTS13 und infolgedessen zu einem beschleunigten Abbau. Zunehmend hält daher die genetische Laboranalytik Einzug in die VWS-Diagnostik (v. a. NGS). Es findet sich jedoch in Teilen eine große Variabilität sowie Komplexität im VWF-Gen – dies auch bei Gesunden [9]. Hilfreich ist die Molekulargenetik aber bei der Diagnostik des Typ 2. Die Mutationen des VWF sind in einer öffentlich zugänglichen ISTH-Datenbank der Sheffield Universität einsehbar.
Therapie
Die Behandlung des VWS ist u. a. abhängig vom VWS-Typ, vom Ausmaß der Blutung, der zugrunde liegenden Klinik, dem Grad der Erkrankung und dem Ansprechen vorausgegangener Therapien. Bei Typ 1 und Typ 2 (Typen 2A, 2M, 2N) ist unter Beachtung möglicher Nebenwirkungen die Gabe von Desmopressin (DDAVP) möglich. Hierdurch kommt es zur Freisetzung von intrinsischem VWF aus dem Endothelzellpool. Bei Typ 2N ist die Gabe unwirksam, bei Typ 2B kontraindiziert. Die Gesamtdauer der Therapie sollte aufgrund einer Tachyphylaxie zwei bis drei Tage nicht überschreiten. Bei Betroffenen, bei denen die Gabe von DDAVP keine Wirkung zeigt oder kontraindiziert ist, aber auch bei Eingriffen mit einem hohen Blutungsrisiko kommen VWF/FVIII-Konzentrate zum Einsatz. Eine Alternative zu den plasmatischen Produkten stellt ein neuerer, rekombinanter humaner VWF dar. An adjuvanten Maßnahmen stehen insbesondere Tranexamsäure als Antifibrinolytikum zur Verfügung, bei Versagen der Standardtherapie auch noch der rekombinante Faktor VIIa. Die VWF-Gentransfer-Methode ist in Entwicklung; sie bedarf jedoch noch einer Weiterentwicklung und Adaption auf den Menschen [10].
