Europäische Verordnung über In-vitro-Diagnostika (IVDR) - Drei Monate nach Geltungsbeginn
Seit Ende Mai gilt die EU-Verordnung über In-vitro-Diagnostika. Obwohl je nach Risikoklasse verlängerte Übergangsfristen gelten, stellt sie die Hersteller vor große Herausforderungen. Die Europäische Datenbank für Medizinprodukte wird erst ab 2026 in vollem Umfang nutzbar sein; bisher wurde noch kein Referenzlaboratorium benannt und es fehlen immer noch Benannte Stellen.
Schlüsselwörter: IVDD, MDR, EUDAMED, VDGH e. V., Konformitätsbewertungsverfahren

Stefanie Giesener ist Head of Quality Management and Regulatory Affairs bei DiaSys Diagnostic Systems GmbH. Sie ist seit 2017 Vorstandsmitglied des Verbands der Diagnostika-Industrie (VDGH e. V.), Vorsitzende der Arbeitsgruppe Zulassung und Registrierung und stellvertretende Vorsitzende des Ausschusses Regulatory Affairs.
Die Europäische Verordnung über In-vitro-Diagnostika (IVDR) ist seit dem 26. Mai 2017 in Kraft und hatte ihren Geltungsbeginn wie ursprünglich angekündigt am 26. Mai 2022. Trotz immenser Verzögerungen bei der Installation der notwendigen Infrastruktur und der Umsetzung der IVDR hielt der Gesetzgeber am Geltungsbeginn fest. Dies war für die betroffenen Hersteller überraschend, denn während die Europäische Kommission zu Beginn der Corona-Pandemie besonnen reagierte und den Geltungsbeginn der Medizinprodukte-Verordnung (MDR) umgehend um ein Jahr auf den 26. Mai 2021 verschob, entschied sie sich bei der IVDR lediglich zu einer Änderungsverordnung und führte verlängerte Übergangsfristen ein, die an die Risikoklasse der Produkte im Januar 2022 angelehnt sind [1].
Somit war der zeitliche Abstand zwischen dem jeweiligen Geltungsbeginn MDR und IVDR von zwei Jahren auf nur ein Jahr geschrumpft. Dies führte zu weiteren Engpässen bei den Benannten Stellen, denn der wohlbedachte Abstand wurde eigentlich dafür benötigt, zunächst den größten Teil der Medizinprodukte konform zur MDR zu erklären, bevor die Benannten Stellen mit den Verfahren für die In-vitro-Diagnostika starteten.
Die Änderungsverordnung für die IVDR führte zu weiteren Verzögerungen, da die Benannten Stellen nun erst noch damit beauftragt waren, ein letztes Mal EG-Konformitätserklärungen im Rahmen der IVD-Richtlinie (IVDD) zu erstellen. Hersteller mit Produkten laut Anhang II der IVDD benötigten neue Zertifikate, um die neuen Übergangsfristen auch ausnutzen zu können.
Wo steht das regulatorische System nun ca. drei Monaten nach Geltungsbeginn und wie ist die Perspektive für die Hersteller von In-vitro-Diagnostika?
Regulatorische Rahmenbedingungen
Inzwischen sind die notwendigen Anpassungen im nationalen Medizinprodukterecht durch das Medizinprodukte-EU-Anpassungsgesetz (MPEUAnpG), das Gesetz zur Änderung des Medizinprodukterecht-Durchführungsgesetzes sowie weiterer Gesetze (MPDG-ÄndG) erfolgt. Da es sich bei der IVDR um eine europäische Verordnung handelt, ist diese im Gegensatz zur damaligen In-vitro-Diagnostika-Richtlinie 98/79/EG unmittelbar in den Mitgliedsstaaten gültig und bedarf nicht der Umsetzung in nationales Recht. Lediglich Anpassungen der nationalen Gesetze sind notwendig [2].
Neben den gesetzlichen Vorgaben werden kontinuierlich weitere Guidance-Dokumente der Medical Device Coordination Group (MDCG) veröffentlicht. Diese sollen helfen, die Auslegung der gesetzlichen Anforderungen zu präzisieren.
Damit steht für die IVDR nun ein sehr umfänglicher Gesetzesrahmen mit einer 204-seitigen EU-Verordnung (englische konsolidierte Fassung) zur Verfügung. Für Deutschland sind weitere 13 angepasste Gesetze und Verwaltungsvorschriften sowie 28 MDCG-Guidance-Dokumente zu beachten, die Hersteller, Behörden und nicht zuletzt die Benannten Stellen lesen, verstehen und umsetzen müssen [3].
Notwendige Infrastruktur
Benannte Stellen
Derzeit haben für die IVDR nur sieben von 22 bzw. nach Austritt des Vereinigten Königreichs aus der EU noch 18 Benannte Stellen den langwierigen Benennungsprozess erfolgreich durchlaufen, die für die IVD-Hersteller unter der IVDD zur Verfügung stehen (Tab. 1).
Tab. 1: Liste der Benannten Stellen (NANDO, New Approach Notified and Designated Organisations) [4].
Body Type | Name | Land | |
|---|---|---|---|
NB 2265 | 3EC International a. s. | Slowakei | |
NB 2797 | BSI Group The Netherlands B. V. | Niederlande | |
NB 0344 | DEKRA Certification B. V. | Niederlande | |
NB 0124 | DEKRA Certification GmbH | Deutschland | |
NB 0459 | GMED SAS | Frankreich | |
NB 0197 | TÜV Rheinland LGA Products GmbH | Deutschland | |
NB 0123 | TÜV SÜD Product Service GmbH | Deutschland | |
Der Benennungsprozess wurde aufgrund der Corona-Pandemie erheblich erschwert, da die Erst-Audits gemäß den EU-Vorschriften auch vor Ort durchgeführt werden mussten. In Deutschland stehen den Herstellern derzeit nur drei Benannte Stellen zur Verfügung; dem stehen allein schon im VDGH-Verband der Diagnostica-Industrie e. V. mehr als 100 Mitgliedsfirmen mit ihren Produkten gegenüber [5].
Während unter der IVDD die Benannten Stellen nur bei den Hochrisikoprodukten in das Konformitätsbewertungsverfahren involviert waren, die mit etwa 8 % aller auf dem Markt befindlichen Produkte einen relativen geringen Anteil ausmachten, kehrt sich das Verhältnis nun um: ca. 80 % der Produkte benötigen eine Benannte Stelle und verbrauchen damit erhebliche Ressourcen [6].
EUDAMED
Die Europäische Datenbank für Medizinprodukte (EUDAMED), die mit dem Beschluss der EU-Kommission (2010/227/EU) [7] zur Verbesserung der Marktüberwachung ins Leben gerufen wurde, steht noch nicht mit all ihren Modulen zur Verfügung. Hersteller können derzeit nur die Registrierung der Wirtschaftsakteure vornehmen; die Module zur Produktregistrierung sowie weitere Module zur Marktüberwachung sind erst nach vollständiger Funktionsfähigkeit der Datenbank nutzbar. Dies bedeutet, dass die Hersteller selbst nun zwar gerade noch rechtzeitig zum Geltungsbeginn der IVDR eine Single Registration Number (SRN) erhielten, jedoch die Registrierung ihrer Produkte nicht vornehmen können. Stattdessen muss die Produktregistrierung nach wie vor in den nationalen Datenbanken erfolgen. Das heißt, dass Hersteller ihre Klasse-A-Produkte ab Geltungsbeginn mit einer Registrierung in der nationalen Datenbank unter die IVDR heben mussten.
Der im Juni veröffentlichte Zeitplan zur Fertigstellung von EUDAMED zeigt, wie lange es noch dauert, bis sie voll funktionsfähig zur Verfügung steht (Tab. 2) [8].
Tab. 2: Zeitplan der Europäischen Kommission für die Fertigstellung der Eudamed (Stand: Juni 2022); MVP = Minimum Viable Product: Das entwickelte System setzt zumindest die Mindestanforderungen der Medizinprodukteverordnung um.
Quartal | Geplante Schritte | |
|---|---|---|
Q4 2023 | Ende der EUDAMED-MVP-Entwicklung für alle sechs Module. | |
Q1–Q2 2024 | Unabhängiges Audit. | |
Q2 2024 | Die Audit-Ergebnisse werden der Medical Devices Coordination Group (MDCG) präsentiert. | |
Q2 2024 | EUDAMED hat nach dem erfolgreichen Audit die volle Funktionsfähigkeit erreicht. Veröffentlichung einer Bekanntmachung der Kommission im Amtsblatt der Europäischen Union (OJEU). Das vollständige EUDAMED-System wird freigegeben (alle 6 Module). | |
Q4 2024 | Ende der sechsmonatigen Übergangsfrist nach Veröffentlichung der Bekanntmachung im OJEU. Die Verwendung von EUDAMED wird in Bezug auf die Verpflichtungen und Anforderungen für Akteure, Vigilanz, Klinische Prüfungen und Leistungsstudien sowie Marktüberwachungsmodule obligatorisch. | |
Q2 2026 | Ende der 24-monatigen Übergangsfrist nach Veröffentlichung der Bekanntmachung im OJEU. Die Verwendung von EUDAMED wird in Bezug auf die Verpflichtungen und Anforderungen für das Unique Device Identification System (UDI/Device) sowie die Module für Benannte Stellen und Zertifikate obligatorisch. | |
Bis dahin müssen die Hersteller die MDCG Guidance 2022-12 [9] befolgen, welche die alternativen Lösungen bis zur vollständigen Funktionalität von EUDAMED beschreibt.
Referenzlaboratorien
Artikel 100 der IVDR sieht vor, dass EU-Referenzlaboratorien benannt werden, um Produkte der Risikoklassen D und bestimmte Produkte der Klasse C auf Einhaltung der Gemeinsamen Spezifikationen zu überprüfen. Bis heute wurde noch kein EU-Referenzlabor benannt. Daher bestehen auch hier nach wie vor Lücken in der regulatorischen Infrastruktur für die zukünftigen Hochrisikoprodukte (z. B. Sars-CoV-2-Tests) sowie Companion Diagnostics (therapiebegleitende Diagnostika), die eine Einbindung der EMA (European Medicines Agency) erfordern. Die Übergangszeit für Klasse-D-Produkte bis zum Mai 2025 ist für die Hochrisikoprodukte die kürzeste, obwohl das Konformitätsbewertungsverfahren das längste ist (16–24 Monate) [10].
Hersteller
Die Hersteller sind gut über die gesetzlichen Veränderungen informiert, da die meisten von ihnen im deutschen Industrieverband VDGH e. V. organisiert sind, der bei allen Fragestellungen in diesem Bereich unterstützt. Somit konnten sich die Hersteller frühzeitig auf die veränderten Rahmenbedingungen einstellen. Es wurden Projekte aufgesetzt, um die Implementierung des neuen Rechtsrahmens voranzutreiben, interne Prozesse anzupassen und wo notwendig neue zu etablieren. Dazu gehörte auch, die Technische Dokumentation der Produkte an die neuen Anforderungen anzupassen und zur Übermittlung an die Benannten Stellen in vollständig digitaler Form zur Verfügung zu stellen.
Je nach Größenordnung des Produktportfolios und dessen Risikoklassen sowie Unternehmensgröße und -struktur mussten unter Umständen zusätzliche personelle Ressourcen geschaffen sowie Investitionen in die Digitalisierung getätigt werden. Gerade kleine und mittlere Unternehmen, die in Deutschland den Hauptanteil der innovativen Industrie ausmachen, hatten damit eine gewaltige Aufgabe vor sich. Das neue Gesetz wurde allerdings seitens der Industrie befürwortet, da die Anerkennung der Selbstzertifizierung beim CE-Kennzeichen im internationalen Umfeld an Ansehen verloren hatte. Dazu hatte wohl vor allem auch der PIP-Skandal um Brustimplantate beigetragen.
Die Hersteller sind für die IVDR grundsätzlich bereit, doch mussten sie mit Besorgnis beobachten, wie andere Medizinproduktehersteller mit der Implementierung und Umsetzung der MDR durch mangelnde Ressourcen bei den Benannten Stellen zeitlich ins Straucheln gerieten. Dies konnte für IVD-Hersteller nicht ohne Folgen bleiben.
Die Änderungsverordnung vom Januar 2022 [11] brachte für die Hersteller zunächst eine gewisse Erleichterung, da nun neue, an die Risikoklasse der Produkte angepasste Übergangsfristen veröffentlicht wurden (Abb. 1).
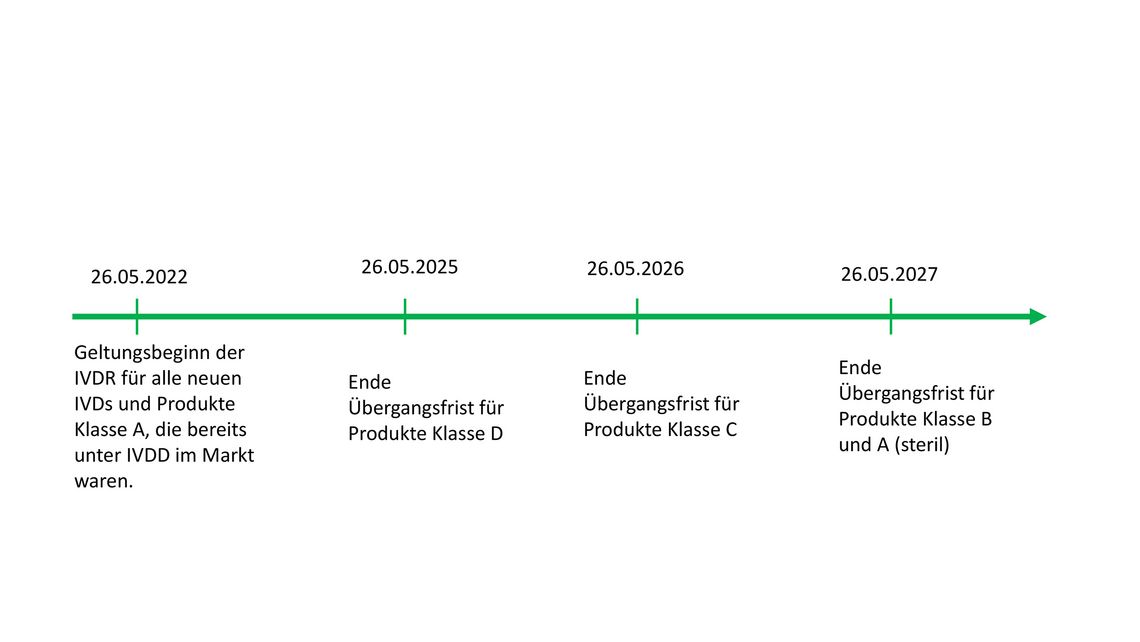
Abb. 1: Übergangsfristen für Produkte verschiedener Risikoklassen.
Dies ist jedoch keine Abhilfe für den eklatanten Mangel an Benannten Stellen. Heute, drei Monate nach Geltungsbeginn, warten immer noch IVD-Hersteller auf ihre Benannte Stelle und können deshalb mit der Konformitätsbewertung ihrer Produkte nicht beginnen.
Verfahren zur Konformitätsbewertung
Diejenigen Hersteller, die sich glücklich schätzen können, eine Benannte Stelle gefunden zu haben, können nun die ersten Konformitätsbewertungsverfahren starten und durchlaufen eine für beide Seiten herausfordernde Lernkurve. Der Erfahrungsaustausch zeigt, dass Hersteller und Benannte Stellen hauptsächlich mit Formalismen zu kämpfen haben. Auch wenn sich beide Seiten wirklich bemühen, dauert es derzeit zwischen zehn und 16 Monaten, bis ein Konformitätsbewertungsverfahren erfolgreich abgeschlossen ist.
Innerhalb dieser Zeit kommt es zu mehreren Reviews der eingereichten Technischen Dokumentation. Der Reviewer der Benannten Stelle muss das Produkt aus den eingereichten Dokumenten verstehen, aber in den meisten Fällen kennt er das Unternehmen und die Produkte vorher nicht. Nach einem ersten Vollständigkeitscheck erfolgt anschließend eine inhaltliche Prüfung mit einem Fragenkatalog. Diesen muss der Hersteller dann beantworten und gegebenenfalls ergänzende Unterlagen einreichen oder bestehende anpassen.
Der Erfahrungsaustausch zeigt, dass die Reviewer sich am Gesetzestext entlang durch die Technische Dokumentation arbeiten und Punkt für Punkt auf Erfüllung prüfen. An dieser Stelle machen sich dann die vielen Redundanzen im Gesetzestext bemerkbar, da es gerade im Bereich der Reports, die der Hersteller für seine Produkte erstellen muss, zu zahlreichen Wiederholungen in den Dokumenten kommt. Hier beginnen dann meist die Diskussionen über Formalismen. Während ein Hersteller eine eher prozessorientierte Dokumentation verfolgt, erwarten die Reviewer eine Dokumentationsstruktur gemäß dem Gesetzestext bzw. der MDCG-Guidance-Dokumente. Diese haben zwar keinen rechtlichen Bindungscharakter, die Benannten Stellen sind aber aufgefordert, diese bei der Konformitätsbewertung zu berücksichtigen.
Letztendlich sind die Hersteller häufig gezwungen, in ihrer Technischen Dokumentation Prozessabläufe zu beschreiben, damit die Reviewer überhaupt verstehen, wie ein Produkt entsteht. Ein Verweis auf interne Verfahrensbeschreibungen als Bestandteil des zertifizierten Qualitätsmanagementsystems, das ohnehin jährlich durch die Benannte Stelle überprüft wird, ist nicht ausreichend. Eine stärkere Vernetzung dieser beiden Systeme wäre wünschenswert, um sich den erheblichen Mehraufwand in der Dokumentation zu ersparen.
Beide Seiten sind bemüht, den Review-Prozess zu verbessern. Es bleibt zu hoffen, dass man aus dem allerersten Konformitätsbewertungsprozess, wenn er denn einmal erfolgreich abschlossen ist, gelernt hat und die folgenden Prozesse dann hoffentlich schneller durchlaufen werden.
Fazit und Ausblick
Die ursprüngliche Zielsetzung, einen soliden, transparenten, berechenbaren und nachhaltigen Rechtsrahmen für In-vitro-Diagnostika zu schaffen, der ein hohes Niveau an Sicherheit und Gesundheitsschutz gewährleistet, gleichzeitig aber innovationsfördernd wirkt, konnte aus Sicht der Hersteller nicht erreicht werden. Im Gegenteil: Der bürokratische Aufwand, mangelnde Ressourcen und die unfertige Infrastruktur für die Umsetzung verlangsamen die Prozesse erheblich und vermeiden Innovation eher, als dass sie sie fördern würden. Zusätzlich besteht die Gefahr, dass wichtige Diagnostika vom Markt genommen werden müssen, wenn trotz verlängerter Übergangsfristen die Produkte nicht rechtzeitig unter die IVDR gehoben werden können. Wenn sich die Anzahl der Benannten Stellen nicht rechtzeitig erhöht, werden auch die verlängerten Übergangsfristen nicht ausreichen, um alle Produkte rechtzeitig unter die IVDR zu heben. Wenn man als IVD-Hersteller schon Bedenken hat, dass es alle Produkte im vorgegebenen Zeitrahmen unter den neuen Rechtsrahmen schaffen, so möchte man sich die Konsequenzen für die potenziellen Patient:innen nicht ausmalen. Zusätzlich muss man sich als deutscher Hersteller fragen, ob zunächst eine Zulassung außerhalb der EU einen schnelleren Marktzugang von Innovationen ermöglicht. Aber was wirft das für ein Bild auf den Standort Europa?