Myelofibrose – neue Aspekte und Strategien
Andrea Kaifie, Steffen Koschmieder
Epidemiologie und Symptomatik
Eine Myelofibrose (MF) kann sowohl de novo entstehen (Primäre Myelofibrose, PMF) oder aus einer Polycythaemia vera (Post-PV-MF) oder einer essenziellen Thrombozythämie (Post-ET-MF) hervorgehen. Die Erkrankungen gehören zu den Philadelphia-Chromosom-negativen myeloproliferativen Neoplasien (MPN) und sind Erkrankungen des fortgeschrittenen Lebensalters. Das mittlere Lebensalter bei Erkrankungsbeginn liegt im Falle der PMF bei 65 Jahren, wobei etwa 11% der Patienten jünger als 46 Jahre sind [1, 2].
Die MF ist eine seltene Erkrankung mit einer Inzidenz von etwa 0,5–1,5 pro 100.000 Einwohner und ist die MPN-Subgruppe mit der geringsten Lebenserwartung [2, 3]. In einer schwedischen epidemiologischen Studie lag die relative 10-Jahres-Überlebensratio der primären Myelofibrose bei 0,21 [4].
Abhängig von der Zuordnung zu einer prognostischen Gruppe liegt die mediane Lebenserwartung zwischen 15,4 Jahren (Niedrigrisiko-Gruppe) und 1,3 Jahren (Hochrisiko-Gruppe, [2]). Zu den häufigsten Todesursachen bei Patienten mit MF gehören maligne hämatologische Erkrankungen (20,1%), gefolgt von kardiovaskulären Erkrankungen (12,3%) und Infektionen (10,4%, [5]).
Während es zu einer Fibrosierung des Knochenmarks kommt, steigt der Grad der extramedullären Hämatopoese in der Milz und in der Leber. Hierbei kann die Milz eine beachtliche Größe erreichen, und das kann zu abdominellen Beschwerden führen.
Neben der Splenomegalie stehen vor allem konstitutionelle Symptome im Vordergrund. Hierzu gehören Fieber, Gewichtsverlust und Nachtschweiß. Weitere Symptome sind Fatigue, Schwindel, Juckreiz, verfrühtes Sättigungsgefühl oder Knochenschmerzen. Weitere Symptome können durch thrombotische/thromboembolische Komplikationen hervorgerufen werden (z. B. Störungen der Mikrozirkulation, Erythromelalgie oder Sehstörungen). Wenn thrombotische/thromboembolische Komplikationen auftreten, geschieht dies meist zu Beginn der Erkrankung in der frühen proliferativen Phase. Später stehen vor allem die durch die insuffiziente Hämatopoese verursachten Symptome im Vordergrund. Hierzu zählen Anämie, Thrombozytopenie und eine erhöhte Neigung zu höhergradigen Infekten aufgrund einer Leukozytopenie [2].
Diagnostik und Molekulargenetik
In Abhängigkeit vom Krankheitsstadium unterscheiden sich die typischen Symptome bei der Erstvorstellung der Patienten. Insbesondere in der Frühphase der Erkrankung, die auch präfibrotische Phase genannt wird, kann eine alleinige Thrombozytose das einzige Symptom der Myelofibrose sein. Hier ist die Abgrenzung zur essenziellen Thrombozythämie (ET) von großer Bedeutung, da sich die Therapie und Prognose von Patienten mit ET meist deutlich von der Therapie und Prognose von Patienten mit MF unterscheiden. Daher muss auch bei diesen Patienten die konsequente Durchführung einer Knochenmark-Diagnostik erfolgen [2].
Gemäß den WHO-Kriterien von 2008 müssen alle folgenden Hauptkriterien und mindestens zwei Nebenkriterien erfüllt sein, um eine primäre Myelofibrose zu diagnostizieren [2, 6].
Hauptkriterien:
• Typische PMF-Knochenmark-Histologie,
• WHO-Kriterien für PV, ET, CML, MDS nicht erfüllt,
• JAK2-Mutation oder MPL515-Mutation oder, falls kein klonaler Marker, kein Hinweis auf sekundäre Myelofibrose.
Nebenkriterien:
• Leukoerythroblastisches Blutbild,
• Erhöhung der Laktatdehydrogenase (LDH),
• Anämie,
• Palpable Splenomegalie.
Für die Post-PV-MF und Post-ET-MF existieren ebenfalls konkrete diagnostische Kriterien, die 2008 von der IWG-MRT erarbeitet wurden. Auch hier kann die Diagnose erst gestellt werden, sobald alle Hauptkriterien und mindestens zwei Nebenkriterien vorliegen [2, 7].
Hauptkriterien:
• Dokumentierte Diagnose einer PV oder ET gemäß den WHO-Kriterien,
• Knochenmark-Fibrose Grad II–III gemäß der europäischen Definition auf einer Sakala von 0–III.
Nebenkriterien:
(Gültigkeit der Nebenkriterien in Klammern):
• Leukoerythroblastisches Blutbild (Post-ET- und Post-PV-MF),
• Palpable Splenomegalie ≥ 5 cm oder neu aufgetretene und palpable Splenomegalie (Post-ET- und Post-PV-MF)
• Neu aufgetretene konstitutionelle Symptome (mindestens eines der folgenden Symptome): über 10% Gewichtsverlust in sechs Monaten, Nachtschweiß, ungeklärtes Fieber > 37,5 °C (Post-ET- und Post-PV-MF),
• Anämie oder nicht mehr erforderliche Aderlass-Therapie in Abwesenheit einer Zytoreduktion (nur Post-PV-MF),
• Anämie oder ein kontinuierlicher Abfall des Hämoglobin-Wertes um ≥ 2 g/dl vom Ausgangswert (nur Post-ET-MF),
• Erhöhte LDH (nur Post-ET-MF).
Bei der körperlichen Untersuchung sollten insbesondere Milz und Leber palpiert werden, ggf. sollte ergänzend eine abdominelle Sonografie zum Ausschluss einer Splenomegalie durchgeführt werden. In der Blutuntersuchung sollten neben Leber-, Nieren- und Gerinnungsparametern auch die Hämolyse-Parameter und der Eisenstatus sowie gegebenenfalls bei Verdacht auf Vorliegen einer systemischen Mastozytose die Serum-Tryptase bestimmt werden. Neben der laborchemischen Diagnostik sollte ein Ausstrich aus dem peripheren Blut zur zytologischen Begutachtung erfolgen. Hier zeigen sich typischerweise ein leukoerythroblastisches Blutbild mit Erythroblasten und einer Linksverschiebung der Granulopoese bis zum Myeloblasten, sowie Tränentropfen-Erythrozyten (Dakryozyten).
Um eine Myelofibrose zu diagnostizieren, ist die Durchführung einer Knochenmark-Punktion essenziell. Prinzipiell sollte sowohl die zytologische als auch die histologische Begutachtung des Knochenmarks erfolgen. Bei einer fortgeschrittenen Fibrosierung ist jedoch häufig die Knochenmark-Aspiration und somit die Auswertung der Zytologie aufgrund einer Punctio sicca nicht möglich. In der Knochenmark-Histologie zeigt sich in der frühen, präfibrotischen Phase eine Vermehrung und Dysplasie der Megakaryozyten. Neben der gesteigerten Megakaryopoese findet sich eine Linksverschiebung der Granulopoese und der Erythropoese. Im Frühstadium zeigt sich häufig noch keine Faservermehrung im Knochenmark. Im fortgeschrittenen Krankheitsstadium zeigt sich dann eine Fibrosierung des Markraums bis hin zur Sklerosierung. Bei unklarer Zuordnung ist die Begutachtung durch einen Referenz-Pathologen zu empfehlen [3].
Wenn Knochenmark aspirabel ist, sollte eine zytogenetische Untersuchung erfolgen. Neben der zytogenetischen Diagnostik ist für die Einordnung des Patienten in eine prognostische Gruppe auch die Molekulargenetik bedeutsam. Diese erfolgt aus dem peripheren Blut und ist somit auch bei Punctio sicca möglich. Die häufigste Mutation ist die JAK2V617F-Mutation. 65% aller PMF-Patienten haben eine JAK2-Mutation, 20 bis 25% der PMF-Patienten eine Calreticulin-Mutation und 6–7% eine MPL-Mutation. Etwa 10–15% aller PMF-Fälle sind „tripel-negativ“, d. h. sie weisen keine der drei oben genannten Mutationen auf [8, 9].
Eine bei der PMF in etwa 20% der Fälle vorkommende Mutation ist die ASXL1-Mutation. Sie ist allerdings nicht spezifisch für MPN, sondern kann auch bei anderen myeloischen Neoplasien vorkommen. Weitere Mutationen (bei über 10% der Patienten) sind solche von TET2, SRSF2 und U2AF1 [9]. Allerdings steigt die Wahrscheinlichkeit, auch als Gesunder eine ASXL1-, TET2- oder DNMT3A-Mutation aufzuweisen, mit dem Alter, von ca. 1% im Alter von 50 Jahren auf bis zu 20% im Alter von 90 Jahren, sodass diese Mutationen bei Patienten mit MPN mit Vorsicht zu beurteilen sind [10–12].
Patienten mit PMF, die eine Calreticulin-Mutation aufweisen, sind jünger, haben einen erhöhten Thrombozyten-Wert, einen niedrigeren DIPPS-plus-Wert und haben weniger häufig eine transfusionsbedürftige Anämie als solche Patienten mit einer JAK2V617F-Mutation [8, 13]. Wird das Gesamtüberleben betrachtet, so zeigen Patienten mit einer Calreticulin-Mutation ein verbessertes Überleben im Vergleich zu Patienten mit einer JAK2-Mutation. Tripel-negative PMF-Patienten und solche, die keine Calreticulin-, aber eine ASXL1-Mutation (CALR-, ASXL1+) haben, haben im Vergleich mit den anderen Gruppen die geringste Lebenserwartung [8]. Tripel-negative Patienten zeigten zusätzlich ein kürzeres Leukämie-freies Überleben. Aufgrund dieser Daten werden tripel-negative und Patienten mit der molekulargenetischen Konstellation CALR-, ASXL1+ als molekulargenetische Hochrisikogruppe eingeordnet [8].
Beurteilung der Prognose von Patienten mit Myelofibrose
Da die allogene Stammzelltransplantation die einzige potenziell kurative Therapiemodalität darstellt, aber mit einer höheren Morbidität und Mortalität einhergeht als die symptomatischen Therapieoptionen, ist es essenziell, die Prognose der Patienten möglichst genau zu bestimmen. Am gebräuchlichsten sind aktuell drei verschiedene prognostische Scores, die im Folgenden beschrieben werden.
IPSS-Score
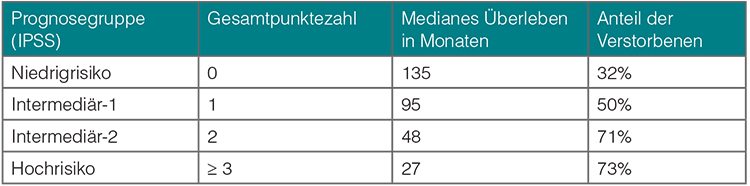
Der IPSS-Score (International Prognostic Scoring System) wurde von der IWG-MRT an einer großen Patientenkohorte (n = 1.054) entwickelt und 2009 von der International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) vorgestellt (Tab. 1, [14]). In diesem Score wird beim Vorhandensein der folgenden Variablen jeweils ein Punkt vergeben:
• Alter > 65 Jahre,
• Konstitutionelle Symptome (Fieber, Gewichtsverlust, Nachtschweiß),
• Hämoglobin ≤ 10 g/dl,
• Leukozyten > 25 G/l,
• Blasten im peripheren Blut ≥ 1%.
Schließlich erfolgt aus der Summe der Punkte zum Zeitpunkt der Erstdiagnose die Zuordnung zu einer Prognosegruppe. Tabelle 1 gibt die mediane Überlebenszeit in Abhängigkeit von der Zugehörigkeit zu einer speziellen Prognosegruppe wieder. In dieser Tabelle wird auch der Anteil der Patienten dargestellt, die während des Studienzeitraums verstorben sind.
DIPSS-Score
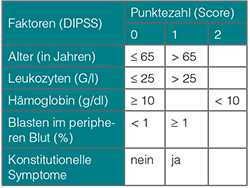
Bei dem „dynamischen“ IPSS (DIPSS) kann auch noch im Krankheitsverlauf die Einteilung der Patienten mit einer Myelofibrose zu einer prognostischen Gruppe erfolgen (Tab. 2, [15]). Hierbei werden die gleichen Variablen wie für den oben beschriebenen IPSS benutzt, allerdings erhält das Vorhandensein einer Anämie mit einem Hämoglobinwert unter 10 g/dl bei diesem Score 2 Punkte.
Auch beim DIPSS werden vier Risikogruppen anhand der Gesamtpunktzahl unterschieden:
• Niedrigrisiko (0 Punkte),
• Intermediär-1 (1–2 Punkte),
• Intermediär-2 (3–4 Punkte),
• Hochrisiko (5–6 Punkte).

DIPPS-Plus-Score
Neben den oben beschriebenen Variablen im DIPSS-Score werden beim DIPSS-Plus-Score weitere Risikofaktoren berücksichtigt (Tab. 3, [16]). Hierzu gehören:
• Transfusionsbedarf,
• Thrombozytenzahl < 100 G/l,
• Ungünstiger Karyotyp (komplex aberrant oder zwei Aberrationen, die -7/7q-, i(17q), inv(3), -5/5q-, 12p- oder 11q23 beinhalten).
Jedem Risikofaktor wird jeweils ein Punkt zugeschrieben, auch einem Hämoglobin-Wert < 10 g/dl (im Gegensatz zum DIPSS). Aus der Gesamtpunktezahl (maximal acht Punkte) der Risikofaktoren werden die Prognosegruppen gebildet (Tab. 3).
Leitliniengerechte Therapie der Myelofibrose
Die Kenntnis der Zugehörigkeit zu einer Prognosegruppe ist notwendig für die weitere Therapieplanung. Neben der krankheitsspezifischen Prognose spielen aber auch die individuellen Begleiterkrankungen eine entscheidende Rolle, um eine für den Patienten passgenaue Therapie zu finden.
Die einzige kurative Behandlungsmöglichkeit ist die Durchführung einer allogenen Stammzelltransplantation. Prinzipiell soll für alle Patienten mit Intermediärrisiko-2- oder Hochrisiko-Konstellation bis zum Alter von 70 Jahren laut Konsensus der EBMT/ELN International Working Group geprüft werden, ob eine allogene Stammzelltransplantation durchführbar ist [17]. Patienten, die ein Intermediärrisiko-1 haben und jünger als 65 Jahre sind und die eine refraktäre, transfusionsabhängige Anämie oder einen ungünstigen Karyotyp aufweisen oder deren Blastenanteil im peripheren Blut > 2% beträgt, sind ebenfalls potenzielle Kandidaten für eine allogene Stammzelltransplantation [17].
Der derzeitige Therapie-Algorithmus der Deutschen Gesellschaft für Hämatologie und Onkologie (DGHO) sieht das in Abb. 1 dargestellte risikoadaptierte Vorgehen vor [2]. Die problemorientierte Therapie im palliativen Arm umfasst Erythropoietin, Erythrozyten-Transfusionen, Hydroxyurea, Interferon, Steroide, Androgene oder IMIDs. Für einen leukämogenen Effekt von Hydroxyurea gibt es bisher keine eindeutige Evidenz [18], er kann jedoch auch nicht gänzlich ausgeschlossen werden.
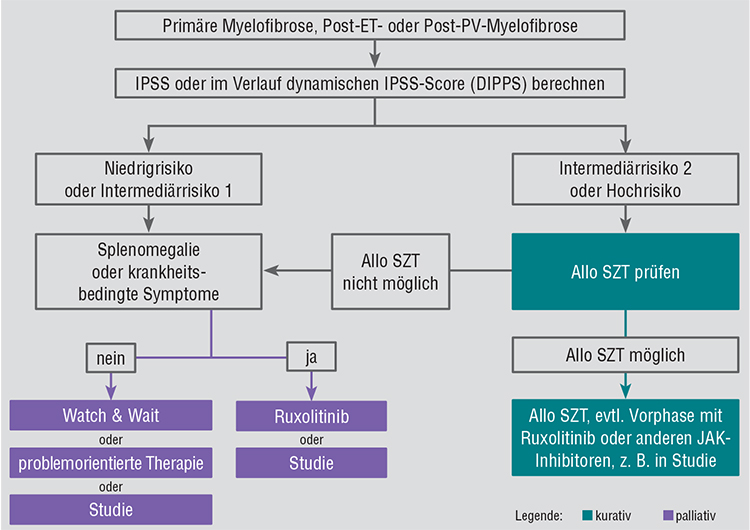
Bei MF-assoziierter Splenomegalie oder krankheitsbedingten Symptomen wird die palliative Gabe von Ruxolitinib oder der Einschluss in eine klinische Studie empfohlen.
Barosi et al. haben untersucht, ob Ruxolitinib einen Effekt auf das Gesamtüberleben bei MF-Patienten hat. Hierfür wurden die COMFORT-Studien hinsichtlich der GRADE-Kriterien (Grading of Recommendation, Assessment, Development, and Evaluation) evaluiert mit dem Schluss, dass diese Studien keine allgemeingültige Aussage in Hinblick auf eine Verlängerung des Überlebens zulassen [19, 20].
Zu den Substanzen, die bei MF-assoziierter Anämie empfohlen werden, gehören Androgene, Prednison, Danazol, Thalidomid oder Lenalidomid. Weitere problemorientierte Therapien bei Patienten mit Niedrigrisiko oder Intermediärrisiko 1 wären Milzbestrahlung oder Splenektomie [3].
Therapieansprechen
Die IWG-MRT hat 2013 die verschiedenen Kategorien des Ansprechens überarbeitet [21]. Alle Kriterien müssen für mindestens zwölf Wochen vorliegen. Nachfolgend werden diese kurz erläutert [2].
1. Komplette Remission (CR)
• Komplettes Ansprechen aller Myelofibrose-assoziierten Symptome inklusive Hepatosplenomegalie.
• Im peripheren Blut ein Hämoglobinwert ≥ 10 g/dl, Thrombozytenzahl ≥ 100 G/l und eine absolute Neutrophilenzahl ≥ 1 G/l.
• Im peripheren Blut fehlender Nachweis von Erythrozyten-Vorstufen, Blasten oder unreifen myeloischen Zellen.
• In der Knochenmark-Histologie Nachweis von Normozellularität,
≤ 5% Blasten und Fibrosegrad ≤ 1.
2. Partielle Remission (PR)
• Im peripheren Blut ein Hämoglobinwert ≥ 10 g/dl, Thrombozytenzahl ≥ 100 G/l, absolute Neutrophilenzahl ≥ 1 G/l und < 2% myeloische Blasten
• UND ein komplettes Ansprechen aller Myelofibrose-assoziierten Symptome inklusive Hepatosplenomegalie, keine extramedulläre Hämatopoese
• ODER in der Knochenmark-Histologie Nachweis von Normozellularität, ≤ 5% Blasten und Fibrosegrad ≤ 1
• UND im peripheren Blut ein Hämoglobinwert ≥ 8,5 g/dl, aber < 10 g/dl, Thrombozytenzahl ≥ 50 G/l, aber < 100 G/l, absolute Neutrophilenzahl ≥ 1 G/l und < 2% unreife myeloische Zellen
• UND ein komplettes Ansprechen aller Myelofibrose-assoziierten Symptome inklusive Hepatosplenomegalie, keine extramedulläre Hämatopoese.
3. Klinische Verbesserung (CI)
3.1 Ansprechen der Anämie
• Anstieg des Hämoglobinwertes um ≥ 2 g/dl bei transfusionsabhängigen Patienten,
• bei Patienten mit einem Hämoglobinwert < 10 g/dl Erreichen einer Transfusionsfreiheit.
3.2 Ansprechen der Splenomegalie
• Milz nicht mehr palpabel, wenn bei der Baseline die Milz zwischen 5 und 10 cm unter dem Rippenbogen tastbar war,
• Milzgrößenreduktion um 50%, wenn bei Baseline die Milz < 10 cm unter dem Rippenbogen tastbar war,
• 35%ige Milzreduktion im CT oder MRT.
3.3 Ansprechen von Symptomen
• 50%ige Reduktion der Gesamtpunktzahl im Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF-TSS) [3].
Neben dem Ansprechen auf eine spezifische Therapie wurden auch Definitionen zur Progression der Myelofibrose, zur stabilen Erkrankung und zum Rezidiv von der IWG-MRT herausgegeben.
Neue Substanzen im Fokus
Im September 2015 haben Tefferi et al. die ersten erfolgsversprechenden Ergebnisse ihrer Pilotstudie mit Imetelstat, einem Telomerase-Inhibitor, publiziert [22]. Von 33 Patienten erreichten unter Imetelstat 21% (n = 7) eine komplette (CR) oder partielle Remission. Bei vier Patienten mit CR kam es zu einer Rückbildung der Fibrosierung des Knochenmarks, und drei dieser Patienten zeigten ein molekulares Ansprechen. Diese Ergebnisse werden derzeit in kontrollierten randomisierten Studien überprüft.
Weitere Substanzen, die derzeit klinisch geprüft werden, sind die JAK-Inhibitoren Momelotinib und Pacritinib. Unter Fedratinib zeigte sich in der klinischen Prüfung ein vermehrtes Auftreten von Wernicke-Enzephalopathien, sodass die Substanz nicht weiterentwickelt wurde [23].
Weitere Substanzen in der klinischen Testung sind mTOR-Inhibitoren, Hedgehog-Signalweg-Inhibitoren, Histondeacetylase-Inhibitoren sowie (pegylierte) Interferone.
Myelofibrose und Lebensqualität
Um die Lebensqualität von Patienten mit myeloproliferativen Neoplasien (MPN) zu beurteilen, finden in der Literatur verschiedene von den Patienten selbst ausgefüllte Fragebögen (Patient-reported Outcomes, PRO) Anwendung. Hierbei werden zum Teil bezüglich der Grunderkrankung unspezifische Lebensqualitäts-Fragebögen genutzt, die generell bei hämatologischen oder onkologischen Patienten verwendet werden können. Beispielsweise wird in dem zweiseitigen EORTC QLQ-C30, einem Fragebogen, der zur Beurteilung der Lebensqualität von onkologischen Patienten dient, neben Fragen zu bestimmten Symptomen auch die generelle Belastbarkeit in alltäglichen Situationen erfragt. Insgesamt werden drei verschiedene Bereiche abgefragt, zur Funktion/Belastbarkeit (fünf Fragen), Symptomen (neun Fragen) und zum allgemeinen Gesundheitszustand (eine Frage; [24]). Zur Differenzierung der Antwortmöglichkeiten werden hier verbalisierte Skalen verwendet (Trifft die Antwort zu: überhaupt nicht, wenig, mäßig, sehr?). Für den EORTC QLQ-C30 existieren noch zusätzliche Entitäten-spezifische Module, z. B. für Mammakarzinom- oder Ovarialkarzinom-Patientinnen.
Neben den unspezifischen Bögen werden zunehmend MPN-spezifische Lebensqualitäts-Fragebögen verwendet, unter anderem, um auch das Ansprechen auf eine Therapie zu dokumentieren. Diese spezifischen Fragebögen beziehen sich auf Symptome, die durch die Grunderkrankung ausgelöst werden und auf den Einfluss, den diese Symptome auf die globale Lebensqualität der Patienten haben. Ein solcher MPN-spezifischer Lebensqualitäts-Fragebogen ist der MPN-SAF (The Myeloproliferative Neoplasm Symptom Assessment Form, [25]). In dem mehrseitigen Fragebogen werden 27 Fragen gestellt, unter anderem zu Fatigue-Symptomen zum Zeitpunkt der Erfassung und während der letzten 24 Stunden. Neben generellen Fragen zur Belastbarkeit werden vor allem MPN-spezifische Symptome (z. B. im Rahmen der Splenomegalie) erfragt. In der letzten Frage wird die allgemeine Lebensqualität der Patienten thematisiert.
Aus dem MPN-SAF wurde der im Umfang deutlich gekürzte einseitige MPN-SAF-TSS (MPN-SAF Total Symptom Score) generiert [26]. In dieser im Umfang reduzierten Variante liegt der Fokus auf denjenigen MPN-Symptomen, die am repräsentativsten für die MPN-Erkrankung sind (z. B. Fatigue, Splenomegalie-assoziierte Symptome, konstitutionelle Symptome).
Sowohl im MPN-SAF als auch im MPN-SAF-TSS werden die Fragen anhand einer Ordinalskala bewertet (0 bis 10). Während der MPN-SAF auch nach der globalen Lebensqualität fragt, wird der MPN-SAF-TSS nach der Summe der einzelnen Antworten ausgewertet, sodass sich ein Wert zwischen 0 und 100 ergeben kann. Der MPN-SAF-TSS wird verwendet, um das Ansprechen auf eine Therapie zu beurteilen. Eine Reduktion von ≥ 50% in dem Fragebogen gilt nach Konsens der IWG-MRT als Ansprechen von Symptomen [3].
Ein rein symptomspezifischer Fragebogen ist sicherlich notwendig, um den Rückgang der durch die Grunderkrankung ausgelösten Symptome zu beurteilen. Wird der Fokus auch auf die globale Lebensqualität gelegt, ist eine reine MPN-spezifische Symptomevaluation nicht mehr ausreichend. Hier sollten künftig weitere Faktoren in die Analyse eingehen, beispielsweise zu sozioökonomischen Determinanten, die neben der soziökonomischen Position (SEP) auch das soziale Gefüge der Patienten und weitere psychoonkologische Parameter beinhalten.
Literatur
1. Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med 2000; 342: 1255-65.
2. Döhner K, Griesshammer M. Aktuelle Therapieoptionen bei der Myelofibrose. Vol. 2. Auflage. 2013, Bremen: UNI-MED Verlag AG.
3. Griesshammer M et al. Primäre Myelofibrose. 2014 14.09.2015]; Available from: www.onkopedia.com/de/onkopedia/guidelines/primaere-myelofibrose-pmf/@@view/html/index.html.
4. Hultcrantz M et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: A population-based study. J Clin Oncol 2012; 30: 2995-3001.
5. Hultcrantz M et al. Risk and cause of death in patients diagnosed with myeloproliferative neoplasms in Sweden between 1973 and 2005: A population-based study. J Clin Oncol 2015; 33: 2288-95.
6. Swerdlow SH et al. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissue. Lyon 2008: IARC.
7. Barosi G et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008; 22: 437-8.
8. Tefferi A et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014; 28: 1472-7.
9. Tefferi A, Pardanani A. Myeloproliferative neoplasms: A contemporary review. JAMA Oncol 2015; 1: 97-105.
10. Busque L et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012; 44: 1179-81.
11. Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371: 2477-87.
12. Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371: 2488-98.
13. Rumi E et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014; 124: 1062-9.
14. Cervantes F et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895-901.
15. Passamonti F et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010; 115: 1703-8.
16. Gangat N et al. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011; 29: 392-7.
17. Kroger NM et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: A consensus process by an EBMT/ELN International Working Group. Leukemia 2015, Aug 21 [Epub ahead of print, DOI 10.1038/leu.2015.233].
18. Bjorkholm M et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011; 29: 2410-5.
19. Barosi G et al. Critical appraisal of the role of ruxolitinib in myeloproliferative neoplasm-associated myelofibrosis. Onco Targets Ther 2015; 8: 1091-102.
20. Harrison C et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012; 366: 787-98.
21. Tefferi A et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood 2013; 122: 1395-8.
22. Tefferi A et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med 2015; 373: 908-19.
23. Pardanani A et al. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J 2015; 5: e335.
24. Aaronson NK et al. The European Organization for Research and Treatment of Cancer QLQ-C30: A quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst 1993; 85: 365-76.
25. Scherber R et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): International prospective validation and reliability trial in 402 patients. Blood 2011; 118: 401-8.
26. Emanuel RM et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: Prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012; 30: 4098-103.

Dr. med. Andrea Kaifie, M. Sc.

Prof. Dr. med. Steffen Koschmieder
Klinik für Hämatologie, Onkologie, Hämostaseologie und Stammzelltransplantation
Medizinische Fakultät der RWTH Aachen