Functional screens for cancer-driving genes: past and emerging tools
Errors in oncogenes and tumor suppressor genes can drive tumorigenesis. Their identification is a priority in cancer research, which can be achieved through functional genomic screens that investigate the relationship between genotype and phenotype. This review discusses how the first cancer driver genes were discovered and introduces the genetic modulating technologies developed since to improve our ability to uncover novel tumorigenic genes. Identifying cancer-driving genes and their role in tumorigenesis is integral for accurate patient diagnosis, tailored treatment regimes and the development of novel therapies.
Schlüsselwörter: Cancer, oncogene, tumor suppressor gene, functional genomics, CRISPR
Genomic mutations arise from both endogenous and exogenous sources with many somatic mutations accumulating over the lifetime of an organism. Not all genomic alterations are detrimental. However, cancer can arise from activating or inactivating mutations in particular genes that provide a proliferative or growth advantage to the cell.
Many cancer-driving genes have been discovered and classified as either oncogenic or tumor-suppressive based on their function in tumorigenesis. While targeted therapies have been developed to treat malignancies defined by some specific genetic alterations, many cancers remain incurable, and others resist therapy or relapse. Understanding tumor driving genes and their deregulated downstream signaling pathways are essential for accurate patient diagnosis, implementation of targeted treatments and accurate anticipation of therapeutic responses – cumulatively giving the best possible outcome for patients.
This review briefly introduces the topic of cancer, the identification of the original cancer-driving genes and the established and emerging technologies being utilized to identify and interrogate novel cancer-driving genes. Cancer researchers can also apply these technologies to understand the intricacies of hallmark pathways, test molecular targets of current therapies and identify novel drug targets.
Introduction
The scientific consensus is that cancer is a genomic disease whereby combinations of genetic alterations affect cellular pathways to drive cancer pathogenesis. Cells accumulate combinations of mutations, including tumorigenic gain-of-function mutations in oncogenes and mostly biallelic loss-of-function mutations in tumor-suppressor genes (TSGs). Over the past century, cancer researchers have steadily unveiled several critical cancer-driving genes to better diagnose, classify and treat numerous cancers.
Despite decades of research on critical genes that contribute to multiple cancers, such as TP53 (mutated in 50 % of all human cancers) and MYC (abnormally expressed in 70 % of all human cancers) [1], we still do not have a complete picture of their role in cancer. For example it is still not well understood what cooperating genes are critical for cancer establishment and maintenance, what downstream cellular pathways are deregulated, how those pathways interact, and how we can design targeted therapies to destroy the cancer.
Researchers are tackling these outstanding questions using an ever-expanding toolbox of technologies. One approach is high throughput deep sequencing of human tumors, which apply statistical analysis to identify commonly mutated putative driver genes. Many sequenced human tumors do not contain mutations in known driver genes, which suggests that many cancer-driving genes are yet to be discovered [2]. An alternate approach is functional genomic screens, which aims to determine the direct impact of genetic alterations on cellular and tumor phenotypes. A significant strength of functional genomic screens is the determination of cancer-driving genes that can be otherwise difficult to pinpoint from background passenger mutations identified by sequencing. The myriad of gene editing tools developed to perform functional genomic screens will be discussed later in this review.
Origins of cancer-driving genes
In 1914, Boveri foreshadowed that cancer growth ensues from incorrect ‘chromosomal’ combinations passed down to daughter cells subsequently causing abnormal cell growth [3]. Surprisingly, this early hypothesis holds if one replaces the word ‘chromosome’ with ‘gene.’ The road to discovering the first cancer-driving gene started in 1911 when Rous studied a transmissible sarcoma in hens [4]. It was not until 1970 that a gene called SRC was identified as the first oncogene. The avian retrovirus Rous Sarcoma Virus (RSV) carried a constitutively active copy of SRC (a gene dispensable for virus replication) to the infected host cells, driving cell proliferation. Viral-SRC lacks the autoregulatory tyrosine present in wildtype cellular SRC that is normally autophosphorylated, thereby rendering it inactive. Other oncogenes such as MYC and RAS were similarly identified from oncogenic viruses [5].
Boveri also predicted that, in addition to overexpression of growth genes, a second category of growth inhibitory genes might be repressed for tumor formation [3]. The first growth-inhibitory gene, now termed tumor suppressor gene, was identified as retinoblastoma (RB) in 1986. This discovery built on Knudson’s work studying children with either hereditary or acquired retinoblastoma. He proposed the “two-hit hypothesis” whereby carriers of a mutated RB gene develop retinoblastoma upon a second ‘hit’ – either another gene mutation or loss of the second RB allele [6]. TP53 was the second tumor suppressor gene identified, having been re-classified from an oncogene (further discussed in 'TP53: A unique tumor suppressor’, see page 106 ff.).
Most of the original cancer-driving genes were discovered from oncogenic viruses or were hereditary and potent drivers of cancer, for example, the BRCA genes in familial breast cancer. To identify cooperating mutations in the context of TP53 mutant cancers, or rare cancer-driving genes, an advancement of technology was required.
Introducing functional genomic screens in cancer
As previously mentioned, functional genomic screens aim to determine how genetic alterations modulate cancer phenotypes. Functional genomic screens are highly customizable depending on the cancer type of interest and the genetic modulation one wishes to induce. A general approach to performing functional genomics (Figure 1) includes delivering a genomic tool to the system that efficiently and robustly alters the expression or function of a gene and hence protein.
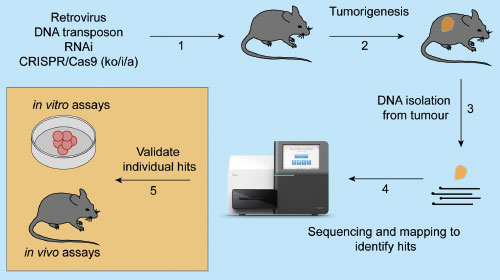
Some of these genetic modifying tools are discussed below. Screens can be performed in vitro or in vivo. However, the system needs to be permissive to the development of tumor formation or appropriate for selecting an altered phenotype such as cell proliferation or cell death [7, 8]. One example is using the Eμ-Myc mouse model that develops fatal B cell lymphomas within ~150 days [9]; cooperating mutations that accelerate cancer progression can be identified in this cancer model. Identifying what altered genes contribute to the phenotype can be achieved through sequencing, such as next-generation sequencing. It is simpler to identify positively selected genetic alterations (enrichment) compared to negatively selected genetic alterations (drop-out). Lastly, multiple candidate driver genes can be identified from one screen, and each hit necessitates individual validation. Further assays can then be performed to interrogate what cellular pathways were deregulated, such as cell proliferation, cell death or metastasis.
Past tools for functional genomics screen in cancer
Insertional mutagenesis screens harnessing oncogenic retroviruses were the first functional genomic screening tools [5]. They capitalize on retroviruses randomly integrating their genetic material into the host cell DNA, thereby causing the deregulation of surrounding genes. Such screens were limited by very restricted cellular tropism, non-random integration patterns and a bias towards activating oncogenes. While there have been significant technical advances in recent years, insertional mutagenesis screens paved the way for making critical discoveries in a high throughput manner [10]. Another earlier developed screening tool utilized sleeping beauty (SB) transposon mutagenesis. Here, a two-part DNA transposon system comprised of a transposase enzyme that ‘cuts and pastes’ a selectable cassette flanked by recognition sequences into the host genome [11]. Both gain-of-function and loss-of-function screens are possible with SB. However, it also exhibits integration bias and off-target overexpression of genes. The lentivirus system, re-engineered from HIV, acts similarly to retroviruses [10]. Unlike retroviruses, lentivirus can infect replicating and non-replicating cells, which provides applications in a wider spectrum of tissues and exhibit a different pattern of integration (albeit also bias towards actively transcribed genes). Altogether, these tools lead to the discovery of many oncogenes important in multiple tissues, but many TSGs were missed [5].
The identification of TSGs became more accessible when RNA interference (RNAi) was established in 2001 [12]. The endogenous pathway that regulates gene expression at the post-transcriptional level in cells was exploited to degrade targeted mRNAs. Short interfering RNA (siRNA)s or short hairpin RNA (shRNA)s were transfected into cells and loaded into the RNA-induced silencing complex (RISC) that promotes degradation of a sequence complementary to cellular mRNAs [13]. This simple tool was revolutionary for loss-of-function screens; however, it had several limitations, including only hypomorphic effects similar to a loss of function rather than knockout, high off-targets and unintended disruption of endogenous degradation of mRNAs. Consequently, these limitations could confound the effect of candidate genes/transcripts pulled out in screens.
Emerging applications of CRISPR/Cas9 in functional genomic screens
The most recently established, and now most widely used, gene-editing tool is the CRISPR/Cas9 system re-engineered from bacteria [14]. The main features of CRISPR technology involve a Cas9 enzyme, being directed by a single guide RNA (sgRNA) to a desired homologous sequence of DNA upstream of a protospacer adjacent motif (PAM) sequence, thereby catalyzing a targeted double-stranded break in the DNA. The host cell's DNA repair machinery is recruited to the DNA double-stranded break and produces insertions or deletions (indels) resulting in a genetic knockout. CRISPR has mostly superseded the previously discussed tools (Table 1) due to its utility and programmability.
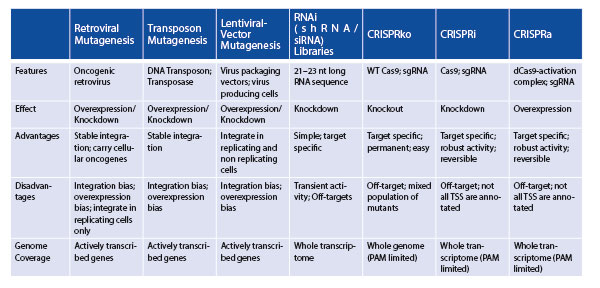
TSGs can be identified in loss-of-function screens with CRISPR knockout (CRISPRko) and through an alternate version of CRISPR capable of transcriptional repression or interference, termed CRISPRi [15]. Alternatively, gain-of-function screens for oncogene discovery can be achieved using CRISPR transcriptional activation (CRISPRa) [16]. CRISPRi and CRISPRa require a modified Cas9, which is enzymatically inactive or dead (dCas9) that is fused to a repressor domain or transcriptional activation machinery, respectively. Unlike CRISPRko, which is directed to intragenic regions, CRISPRi and CRISPRa are recruited to the transcriptional start site of genes by sgRNAs to modulate either activation or repression of target gene expression [13].
Identification of many cancer driving genes can be achieved by high-throughput and systematic CRISPR screens. Here, pooled sgRNA libraries are constructed targeting subsets of genes in certain pathways or, recently, the whole genome. The sgRNA library (containing multiple sgRNAs per gene) and Cas9 are packaged and delivered to target cells/tissues. Lentivirus is a popular delivery vector and this approach gives robust and reproducible gene perturbations resulting in phenotypic readouts in many diverse tissue types.
Conclusions
Several cancer-driving genes have been uncovered, but sequencing data suggests more are yet to be found and therefore therapeutically targetted. The advancement of gene editing tools to perform functional genomic screens has exponentially improved our ability to identify novel cancer-driving genes. One caveat to these technologies is that not all genetic alterations can be recapitulated using these tools. Single nucleotide variants make up a large proportion of genetic alterations in cancer and can produce altered protein function rather than a simple loss-of-function or constitutive overexpression [17]. Mutant TP53 is an example of altered protein function arising from point mutations in the DNA binding domain that may alter regulation of downstream pathways compared to normal [18].
Development of high throughput gene-editing tools capable of producing specific point mutations would allow investigation of altered protein functions in cancer. A tool of this nature has recently been developed named CRISPR base editing. The Liu group fused a deaminating enzyme to dCas9, which creates an “editor” that can change a base from either C to T or A to G [19]. This tool is very recent, and its editing activity was initially performed in cell lines. However, there is limited evidence showing its activity in different primary cell types. Further optimization of base editing or another tool of this nature would allow better recapitulation of cancer biology. This includes simulating the order of mutation acquisition as cancer progresses and responds to therapy.
Other than the identification of novel oncogenes and TSGs, functional genomic screens can be applied to many aspects of cancer research. For example, screening for synthetic lethality (where certain combinations of genetic mutations result in cell death) and cancer immunotherapy targets or effectors. Moreover, mutations that confer resistance to therapies can be screened [20]. Altogether, these insights into cancer biology using functional genomics will improve patient outcomes through better diagnosis, targeted cancer therapy, novel combination therapies and development of new drugs.
Autoren
